2023
05-07
05-07
速看!单细胞测序分析流程解析来了 | 单细胞专题
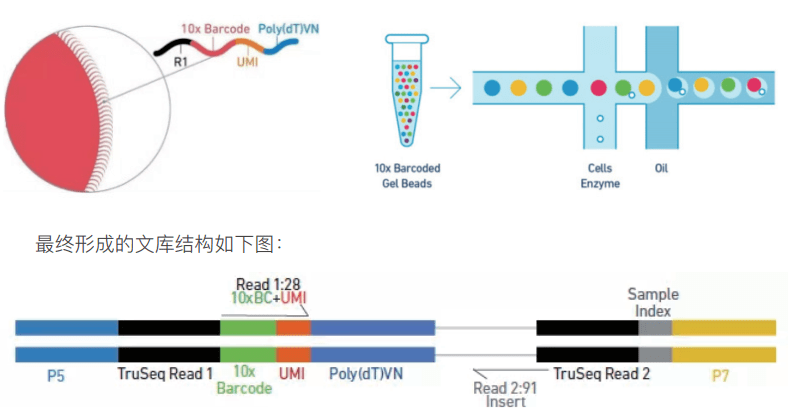 10x genomic 平台可以一次捕获 1000~10000 个细胞,对于 v3 试剂,要求每个细胞至少测 20k reads,按照测序模式 PE150 进行计算,当捕获 5000 个细胞时,最少需要的数据量为 =5000×20000(reads)×300(bp)=30×109,即捕获 5000 个细胞时,至少要测 30G 的数据量。随着捕获细胞数的增加,测序量也是成倍增加,如何...阅读全文&... 阅 读 全 部 >
10x genomic 平台可以一次捕获 1000~10000 个细胞,对于 v3 试剂,要求每个细胞至少测 20k reads,按照测序模式 PE150 进行计算,当捕获 5000 个细胞时,最少需要的数据量为 =5000×20000(reads)×300(bp)=30×109,即捕获 5000 个细胞时,至少要测 30G 的数据量。随着捕获细胞数的增加,测序量也是成倍增加,如何...阅读全文&... 阅 读 全 部 >
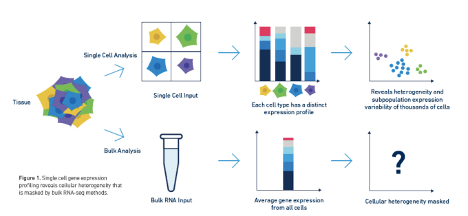 超越转录组:单细胞表观基因组学的价值?长期以来,研究人员一直在基于转录组数据来探索有关复杂细胞群体和过程的生物学见解。样本本身的异质性以及基因表达的细胞间差异促使科学家从批量(bulk)细胞的平均值测定转向单细胞RNA测序,以便更准确地描述复杂的生物系统。然而,单单凭借转录组的信息,也许无法讲述细胞的完整故事,科学家通常还想了解造成这些基因表达差异背后的原因 – 也就是说,从机理上解释通...阅读...
超越转录组:单细胞表观基因组学的价值?长期以来,研究人员一直在基于转录组数据来探索有关复杂细胞群体和过程的生物学见解。样本本身的异质性以及基因表达的细胞间差异促使科学家从批量(bulk)细胞的平均值测定转向单细胞RNA测序,以便更准确地描述复杂的生物系统。然而,单单凭借转录组的信息,也许无法讲述细胞的完整故事,科学家通常还想了解造成这些基因表达差异背后的原因 – 也就是说,从机理上解释通...阅读... 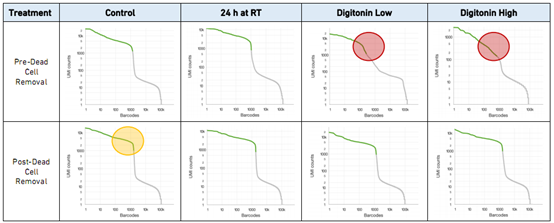 1为什么要去死细胞10× Genomics单细胞方案要求使用具有较高活性的单细胞悬液。在实际实验中,因为样本类型和制备单细胞悬液方法的不同,容易出现单细胞悬液中死亡细胞的比例较高的情况。去除单细胞悬液中的死细胞和其他污染物对获得高质量数据是至关重要的。这是因为,死亡的细胞易裂解导致其中的RNA释放出来。这种cell-free RNA会导致检测的背景噪声,并会影响单细胞数据的质量。下文我们会...阅...
1为什么要去死细胞10× Genomics单细胞方案要求使用具有较高活性的单细胞悬液。在实际实验中,因为样本类型和制备单细胞悬液方法的不同,容易出现单细胞悬液中死亡细胞的比例较高的情况。去除单细胞悬液中的死细胞和其他污染物对获得高质量数据是至关重要的。这是因为,死亡的细胞易裂解导致其中的RNA释放出来。这种cell-free RNA会导致检测的背景噪声,并会影响单细胞数据的质量。下文我们会...阅... 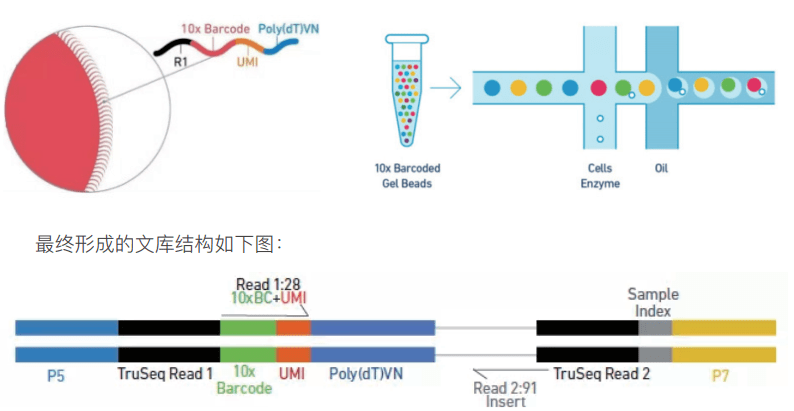 10x genomic 平台可以一次捕获 1000~10000 个细胞,对于 v3 试剂,要求每个细胞至少测 20k reads,按照测序模式 PE150 进行计算,当捕获 5000 个细胞时,最少需要的数据量为 =5000×20000(reads)×300(bp)=30×109,即捕获 5000 个细胞时,至少要测 30G 的数据量。随着捕获细胞数的增加,测序量也是成倍增加,如何...阅读全文&...
10x genomic 平台可以一次捕获 1000~10000 个细胞,对于 v3 试剂,要求每个细胞至少测 20k reads,按照测序模式 PE150 进行计算,当捕获 5000 个细胞时,最少需要的数据量为 =5000×20000(reads)×300(bp)=30×109,即捕获 5000 个细胞时,至少要测 30G 的数据量。随着捕获细胞数的增加,测序量也是成倍增加,如何...阅读全文&...  不论是做实验还是做高通量测序分析,我们往往离不开要对分子层面的作用机制进行研究,而其中基因可以编码下游蛋白,通过调控蛋白之间的相互作用或是影响蛋白质本身行使的功能从而最终影响表型的变化。这么一来,对于蛋白作用的了解就显得格外重要了。这些信息可以来源于KEGG数据库的注释也可来源于Uniprot对蛋白功能的注释,但不论是存在于KEGG还是Uniprot数据库的信息,都过于分散,是否有已经整理好的蛋白...
不论是做实验还是做高通量测序分析,我们往往离不开要对分子层面的作用机制进行研究,而其中基因可以编码下游蛋白,通过调控蛋白之间的相互作用或是影响蛋白质本身行使的功能从而最终影响表型的变化。这么一来,对于蛋白作用的了解就显得格外重要了。这些信息可以来源于KEGG数据库的注释也可来源于Uniprot对蛋白功能的注释,但不论是存在于KEGG还是Uniprot数据库的信息,都过于分散,是否有已经整理好的蛋白... 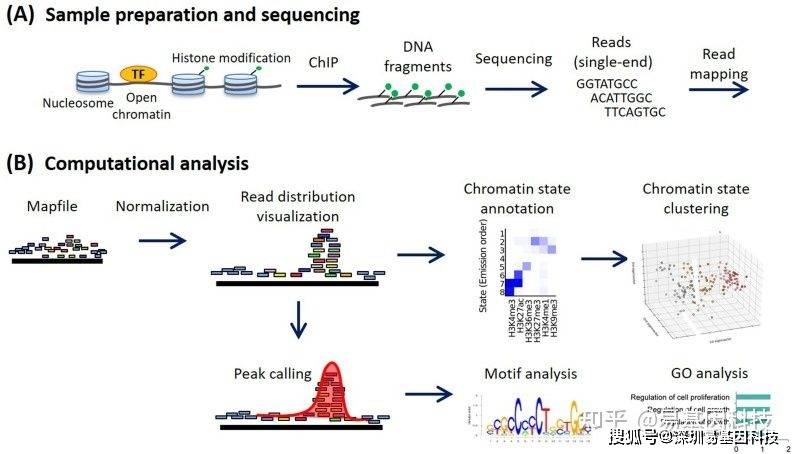 大家好,这是专注表观组学十余年,领跑多组学科研服务的易基因。2020年03月,《Methods》杂志上发表一篇关于表观组学ChIP-seq分析方法的综述文章,详细介绍了染色质免疫共沉淀(ChIP-seq)的工作流程和高级应用。以下为原文总结分享:一、介绍(Introduction)染色质免疫共沉淀测序(ChIP-seq)是表观基因组学研究中的一种主要方法。全基因组的组蛋白...阅读全文>&g...
大家好,这是专注表观组学十余年,领跑多组学科研服务的易基因。2020年03月,《Methods》杂志上发表一篇关于表观组学ChIP-seq分析方法的综述文章,详细介绍了染色质免疫共沉淀(ChIP-seq)的工作流程和高级应用。以下为原文总结分享:一、介绍(Introduction)染色质免疫共沉淀测序(ChIP-seq)是表观基因组学研究中的一种主要方法。全基因组的组蛋白...阅读全文>&g...  大规模并行测序技术或下一代测序已成为基因诊断和研究的标准技术,尤其是外显子组和基因组测序现在已经在世界范围内广泛应用于患者的分子诊断。在过去几年中,许多实验室都在努力应对基于全新技术建立基因检测工作流程的挑战。测序技术中持续引入新的仪器、化学和分析方法加剧了这些挑战。在过去十年中,新的测序技术已经上市,而其他技术已经消失,并且所有这些技术都经历了快速的变化和升级。外显子组捕获试剂盒、配套设备和耗材...
大规模并行测序技术或下一代测序已成为基因诊断和研究的标准技术,尤其是外显子组和基因组测序现在已经在世界范围内广泛应用于患者的分子诊断。在过去几年中,许多实验室都在努力应对基于全新技术建立基因检测工作流程的挑战。测序技术中持续引入新的仪器、化学和分析方法加剧了这些挑战。在过去十年中,新的测序技术已经上市,而其他技术已经消失,并且所有这些技术都经历了快速的变化和升级。外显子组捕获试剂盒、配套设备和耗材...  大规模并行测序技术或下一代测序已成为基因诊断和研究的标准技术,尤其是外显子组和基因组测序现在已经在世界范围内广泛应用于患者的分子诊断。在过去几年中,许多实验室都在努力应对基于全新技术建立基因检测工作流程的挑战。测序技术中持续引入新的仪器、化学和分析方法加剧了这些挑战。在过去十年中,新的测序技术已经上市,而其他技术已经消失,并且所有这些技术都经历了快速的变化和升级。外显子组捕获试剂盒、配套设备和耗材...
大规模并行测序技术或下一代测序已成为基因诊断和研究的标准技术,尤其是外显子组和基因组测序现在已经在世界范围内广泛应用于患者的分子诊断。在过去几年中,许多实验室都在努力应对基于全新技术建立基因检测工作流程的挑战。测序技术中持续引入新的仪器、化学和分析方法加剧了这些挑战。在过去十年中,新的测序技术已经上市,而其他技术已经消失,并且所有这些技术都经历了快速的变化和升级。外显子组捕获试剂盒、配套设备和耗材...  基因Panel 常称之为“基因包”,也可以称之为“基因组合”,商业化的市场习惯称之为“基因套餐”。就 基因Panel 本身来说,其实并没有指明所检测的基因数目应该是多少。几个基因可以是一个 Panel,几十个基因也可以是一个 Panel,几百成千上万个基因也可以是一个 Panel。就 Panel大小来说,不仅需要看检测基因的数目,还需要看基因所覆盖的区域大小。经过近百年的努力直至近年来...阅读全...
基因Panel 常称之为“基因包”,也可以称之为“基因组合”,商业化的市场习惯称之为“基因套餐”。就 基因Panel 本身来说,其实并没有指明所检测的基因数目应该是多少。几个基因可以是一个 Panel,几十个基因也可以是一个 Panel,几百成千上万个基因也可以是一个 Panel。就 Panel大小来说,不仅需要看检测基因的数目,还需要看基因所覆盖的区域大小。经过近百年的努力直至近年来...阅读全...  全外显子(Whole-exome sequencing)测序是啥?转录组(RNA-seq)测序是啥?ChIP-seq又是啥?它们之间有什么差别么?傻傻分不清,不用怕,多学习下就会了,下面让我们一起来从平均测序深度和区域覆盖度的角度来区分它们吧!1 基础概念平均测序深度:指定区域内得到的所有碱基数目与该区域的长度的比值,如果是全基因组,就是整个测序的碱基数目...阅读全文>>...
全外显子(Whole-exome sequencing)测序是啥?转录组(RNA-seq)测序是啥?ChIP-seq又是啥?它们之间有什么差别么?傻傻分不清,不用怕,多学习下就会了,下面让我们一起来从平均测序深度和区域覆盖度的角度来区分它们吧!1 基础概念平均测序深度:指定区域内得到的所有碱基数目与该区域的长度的比值,如果是全基因组,就是整个测序的碱基数目...阅读全文>>... 