10x genomic 平台可以一次捕获 1000~10000 个细胞,对于 v3 试剂,要求每个细胞至少测 20k reads,按照测序模式 PE150 进行计算,当捕获 5000 个细胞时,最少需要的数据量为 =5000×20000(reads)×300(bp)=30×109,即捕获 5000 个细胞时,至少要测 30G 的数据量。随着捕获细胞数的增加,测序量也是成倍增加,如何从庞大测序数据中获得单个细胞的表达谱呢?下面为您介绍 10X 单细胞转录组测序分析流程及原理。
01
10x 单细胞标记原理
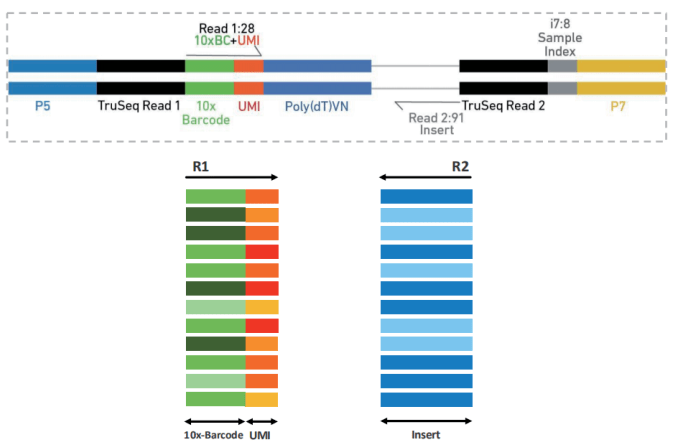
在介绍分析流程之前,先简单回顾下 10x 单细胞标记原理以及文库结构,便于对后续分析的理解。Gel bead(凝胶磁珠,下图左)上含有全长 Illumina TruSeq Read 1 测序引物、16nt 10X Barcode 序列、12nt unique molecular identifier (UMI)、30 nt poly dT 反转录引物。将制备好的单细胞悬液,Gel bead 和油滴,经由微流体“双十字”交叉系统形成 GEM(即油包水结构,下图右)。
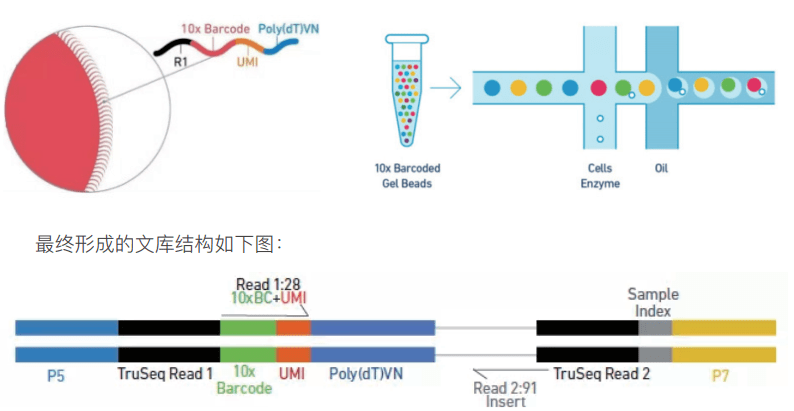
前面我们已经提到过按照捕获 5000 个细胞,至少需要测 30G 的数据量,一般情况下,测序数量要按照每个细胞测 30-50k reads 进行计算,对应的数据量为 45G-75G。跟普通转录组(一般测序数据量 6G)相比,测序数据量非常庞大。因此,为了降低测序时Sample Index 读取的偏好性,每个 Sample Index 含有四种 8 碱基长度的序列(如下图)。
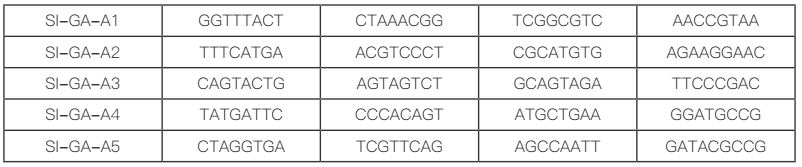
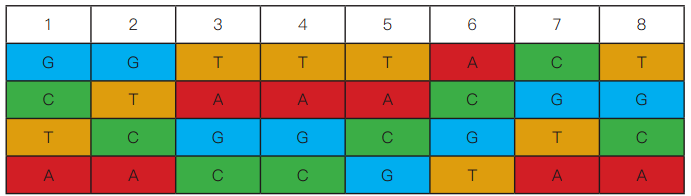
以 SI-GA-A1 为例,对应的四种 index 信息如下图,四种 index 保证了每个位置上都包含了 A,T,C,G 四种碱基,从而消除了某个位置碱基的不平衡性。因此,在数据拆分时,同一样本下机数据会按照 4 种 Sample index 分成四份,在数据分析前,需要将 4 份数据进行合并。
02
10x 单细胞分析流程
拿到下机数据后,如何获得单个细胞的表达谱呢?首先,利用 10x Genomics 提供 的 官 方 分 析 软 件 CellRanger(https://support.10xgenomics.com/single-cell-geneexpression/software/overview/welcome) 对原始数据进行数据质控过滤、比对、定量、鉴定回收细胞,最终得到各细胞的基因表达矩阵。后续采用 Seurat(https://satijalab.org/seurat/)进行进一步的细胞过滤、标准化、细胞亚群分类、各亚群差异表达基因分析及 Marker 基因筛选。基本分析流程图如下:
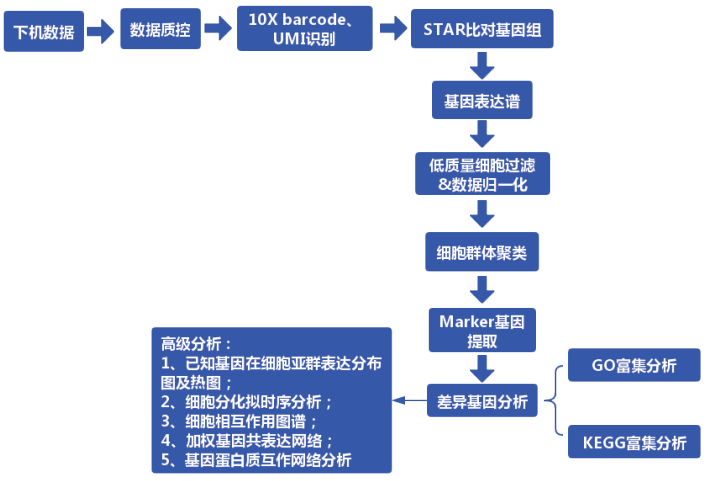
具体的分析步骤为:
细胞 barcode,UMI 提取
提取R1端的10x Barcode(16bp,用于区分细胞)和UMI序列(12bp,用于基因定量),以及 R2 端的插入片段序列(用于基因比对,R2 端序列官方推荐长度:v3 试剂 91bp,v2试剂 98bp)。
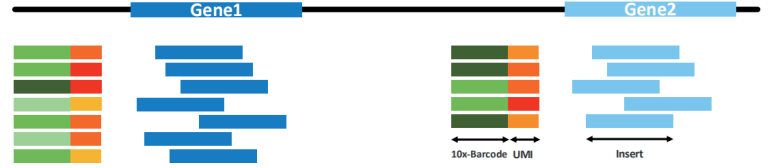
比对
使用 STAR (Spliced Trans Alignment to a Reference) 软件将 reads(上步提取的 91bp 的 R2 插入片段)比对到参考基因组上,使用基因组 GTF 注释文件进行校正,区分出外显子区、内含子区、基因间区。具体的区分规则为:至少 50% 比对到外显子区的reads 记为外显子区,比对到非外显子区且与内含子区有交集的 reads 记为内含子区,其他的均为基因间区。
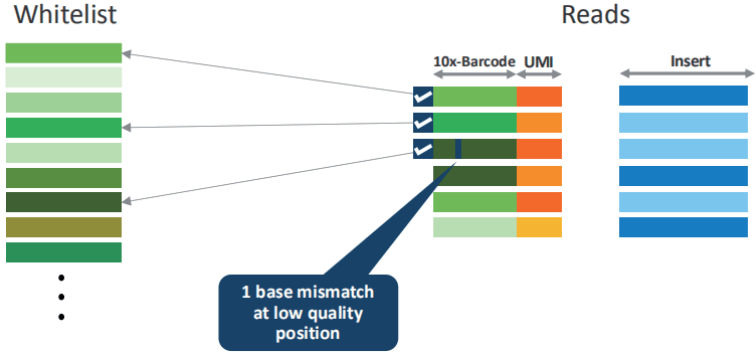
barcode 校正
将测序获得的 Barcode 序列信息与数据库中已知 Barcode 序列进行比对,与数据库中已知 Barcode 完全一致的 barcode 序列(测得的序列)为真实的序列。如果比对时有一个碱基发生错配且该位置碱基质量值比较低时会按照已知的序列进行校正,其余情况的barcode 为无效的,后续分析时直接舍弃。
UMI 过滤和校正
测序得到的 UMI 序列并不是简单的直接用于后续的分析,还需要对不合格的 reads 进行过滤及校正。具体过滤及校正方法如下:
UMI 需满足以下条件:
1. 必须不能是同聚物,比如 AAAAAAAAAA;
2. 必须不能含有 N 碱基;
3. 必须不能包含质量值小于 10 的碱基;
UMI 校正:
具有相同 barcode 的 UMI,如果只相差一个碱基的 mismatch,会按照数目多的 UMI碱基序列进行矫正。如下图 C 被校正为 G。

UMI 计数
对所有的有效 barcode 进行计数(注意:只有包含有效的 barcode 和 UMI 且可靠比对的 read 才能用于计数),获得未进行过滤的原始细胞基因表达矩阵。

call cells
细胞捕获时,一次捕获可产生近百万的 GEMs(GEMs 如下图最左侧),这些 GEMs可能存在以下几种情况:
A. 一小部分 GEM 包含一个 cell(下图红色示例图);
B. 很少部分 GEM 会包含多个 cells(下图绿色示例图);
C. 很大部分 GEM 是空的,不包含任何细胞,但含有背景 RNA,这些细胞被称为背景barcode(下图黄色示例图);

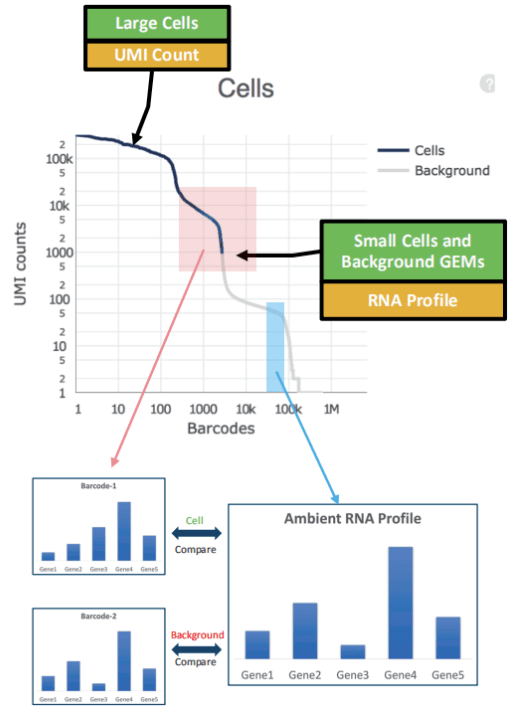
以上步骤均有 10x 官方软件 CellRanger 完成,在 call cells 的步骤中,我们发现有一些 GEM 会包含多个 cells,这部分细胞在鉴定时由于 UMI 表达量比较高,会在 Step1 时就被检出,但实际上这部分并非单个细胞。此外,还有一些部分细胞可能处于凋亡状态,在实际检测出可能检测到的基因数量偏低以及线粒体基因大量表达。对于这些多细胞数据或者低质量的细胞(细胞处于凋亡状态或者细胞碎片等),CellRanger 无法直接检测出,还需要依赖第三方软件 Seurat 对细胞进行过滤。
细胞过滤
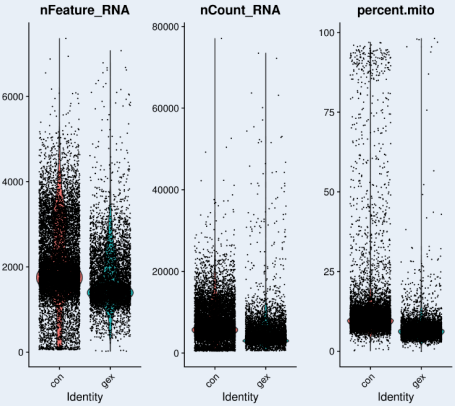
Seurat 可以导入 CellRanger 生成的表达谱文件,除了统计出每个细胞对应的基因数目,UMI 数目,还可以计算出每个细胞对应的线粒体基因表达情况等。通常将基因数目小于 500 的细胞(当然,如果研究的目标细胞中含有中性粒细胞或者前期未进行裂红处理含有大量红细胞等 RNA 含量较低的细胞,可以适当调整该阈值),线粒体比例超过一定比例的细胞进行过滤,具体采用哪个条件进行细胞过滤,不同的组织不同的项目有不同的参数,需要根据项目的基本情况后期再进行调整。比如以下的数据,我们可以按照 nFeature-500-4000(纵坐标为细胞对应的 gene 数量,每个点代表一个细胞),nCount-500-20000(纵坐标为细胞对应的 UMI 值),percent.mito- 线粒体比例按照<25% 进行细胞过滤。
二次分析(降维,聚类,差异等分析)
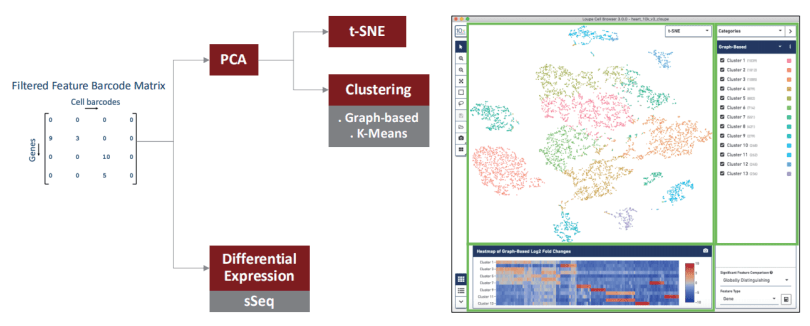
基于上一步得到的细胞基因表达谱,通过降维(PCA),聚类(Graph-based & K-Means)以及可视化(t-SNE)得到常见的细胞聚类结果,输出的结果可以用配套的 Loupe CellBrowser 软件进一步细致的研究。
过滤后细胞聚类及差异分析
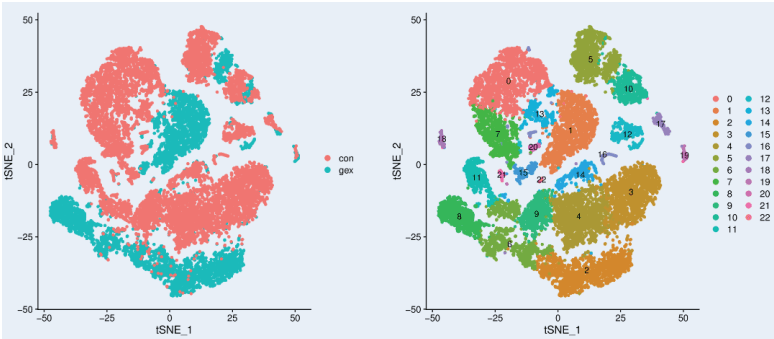
基于过滤后获得的高质量的细胞基因表达谱,对细胞进行聚类分析,获得各细胞对应的 cluster 信息。并对每个 cluster 细胞与其他细胞之间进行差异分析,获得在该类细胞中显著性差异上调的基因,以及差异基因的 KEGG/GO 富集分析。
到这里,单细胞的基本分析就完成了,当然这里只是获得了一些基础的信息,后续仍需要深入的数据挖掘及多样的个性化分析来更好的解释生物学问题。
转自:联川生物
- 本文固定链接: https://maimengkong.com/1504.html
- 转载请注明: : 萌小白 2023年5月1日 于 卖萌控的博客 发表
- 百度已收录
