大家好,这是专注表观组学十余年,领跑多组学科研服务的易基因。
2020年03月,《Methods》杂志上发表一篇关于表观组学ChIP-seq分析方法的综述文章,详细介绍了染色质免疫共沉淀(ChIP-seq)的工作流程和高级应用。以下为原文总结分享:
一、介绍(Introduction)
染色质免疫共沉淀测序(ChIP-seq)是表观基因组学研究中的一种主要方法。全基因组的组蛋白修饰分析(如增强子分析和全基因组染色质状态注释)可以系统分析表观基因组如何促进细胞身份(cell identity)、细胞发育、细胞谱系规范(lineage specification)和疾病。本综述中,作者专注于生物学研究的实践方法,首先介绍了ChIP-seq从质量评估到染色质状态注释的标准分析工作流程。接下来作者概述了几种用于组蛋白修饰的ChIP-seq高级应用,包括预测基因表达水平、染色质成环 (enhancer-promoter looping)、数据归集(data imputation)。最后,作者讨论了单细胞ChIP-seq(scChIP-seq)分析方法,阐明了复杂组织和癌症中的细胞多样性。
Roadmap Epigenomics Consortium提出的五个"主要组蛋白标记"广泛用于 ChIP-seq 分析:
- H3K4me1和H3K27ac:与增强子区域相关;H3K4me3:与启动子区域相关;H3K36me3:与基因体中的转录区域相关;H3K27me3:与 Polycomb 抑制相关;H3K9me3:与异染色质相关。
二、ChIP-seq实验和分析工作流程
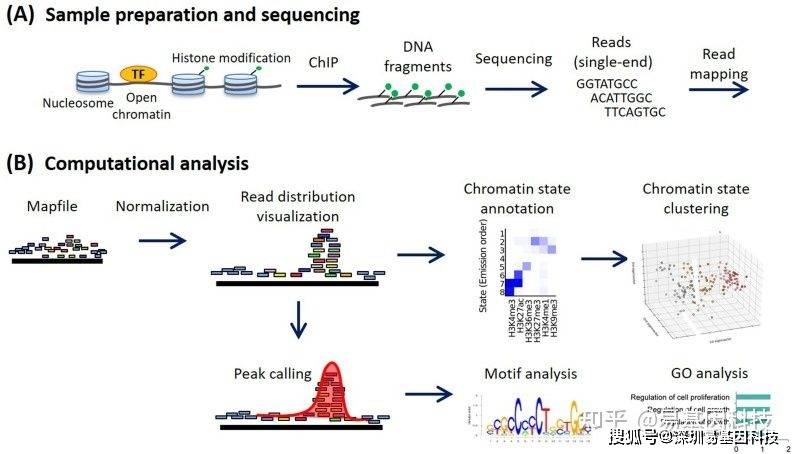
图1:ChIP-seq实验和分析工作流程。(A)样品制备和测序;(B)ChIP-seq标准分析流程。
(1)环境设置(Environmental setup)
NGS分析的计算工具通过各种计算机语言编写,例如C++,R,Python,Java和Perl语言。每种语言都需要不同的设置方法。大多数在Linux系统上执行,也可以使用Linux的Mac终端和Windows Subsystem for Linux (WSL)。Python2在2020年以后不再维护,有几种工具需要Python2但尚未更新到Python3,因此用户不得不考虑更换软件包以获得新的替代方案。如果想继续使用旧的应用程序,可以选择Docker、Singularity提供安全、奇异(https://sylabs.io/)的分析环境。
(2)从公共数据库中下载ChIP-seq数据
多个公共数据库可以下载组蛋白修饰ChIP-seq数据,如人内皮细胞表观基因组数据库,包含人体9种血管类型中获得的424个组蛋白修饰ChIP-seq和67个RNA-seq数据集,有包括reads、比对文件、bigwig文件和peaks表等多种数据类型可用,这些数据适合用作ChIP-seq分析的教学和测试数据(表1)。
 表1: ChIP-seq公共数据库
表1: ChIP-seq公共数据库
(3)组蛋白修饰ChIP-seq分析的技术考虑
ChIP-seq分析的可靠性取决于抗体质量,包括特异性和信噪比(S/N)。非特异性抗体-DNA结合的假阳性富集位点可能会干扰分析,因此应使用多种抗体验证ChIP-seq结果。
虽然大多数ChIP-seq工具都是针对特定基因组区域的sharp peaks,如转录起始位点(TSS),但一些组蛋白修饰与大基因组结构域相关,从而导致富集区域广泛分布。H3K27me3和H3K36me3富集分布在几百个碱基上,而H3K9me3 peaks通常扩展到几兆碱基。增强子标记H3K27ac和H3K4me1产生sharp peaks,但有时也会构建broad富集区域,称为“超级增强子”。H3K4me3启动子标记还可以覆盖小鼠卵母细胞中的broad结构域。这种peak形状和宽度变化影响最佳计算工具的选择。比如,ROSE用于检测超级增强子位点,Music用于计算要研究样本平均的peaks宽度。
(4)reads比对
使用Bowtie、Bowtie2或BWA等工具对测序reads(FASTQ或CSFSATQ格式)进行比对。Bowtie2和BWA可以通过gapped alignments比对考虑INDEL(insertions和deletions),这适用于长reads和双端reads。比对文件有多种格式,如SAM、BAM、CRAM和tagAlign,BAM格式是迄今为止使用最多格式,不过更节省空间的CRAM格式正在成熟,很可能成为下一个标准。比对后,比对到相同基因组位置的reads被过滤为冗余reads,去冗余后剩余的reads用于后续分析。
(5)Peak calling
peak-calling可以鉴定基因组中显著富集位点(peaks)。peak-calling结果通常以BED格式呈现。尽管ChIP-seq peaks没有strand信息,但可以从基因信息中预测(如关注TSS周围富集的组蛋白标记)。MACS2是最常用的peak-calling工具,不过没有任何工具可以达到100%准确度。因此,一种实用的策略是采用较为宽松的阈值获得大量包含真实阳性信号和干扰信号的peaks,然后使用另一种方法进一步提取以提高特异性(如使用不重复的发现率(IDR))在生物学重复中选择一致的信号。
(6)ChIP-seq 数据质量评估
ChIP-seq样品的质控(QC)对于判断测序数据是否高质量且能否进一步分析至关重要。其中特别重要的指标:
- 比对率。反映reads质量和基因组DNA的测序reads比例。reads深度(去冗余后比对的reads数量)。足够的reads深度取决于基因组大小和抗体信噪比。ENCODE consortium建议至少10M唯一比对的reads作为分析人样本的sharp-mode peaks最小值。broad组蛋白标记通常具有较弱的信噪比,且需要更多的reads(人样本>40M)作为peak-calling的最小值。文库复杂性(非冗余reads比例)。范围从0到1,ENCODE认为10M比对reads的复杂性应 > 0.8。归一化strand系数(NSC,通过SSP计算)。sharp和broad peaks的信噪比(S/N)指标,使用10M ChIP-seq公共数据库对多个物种进行深入验证,建议阈值为NSC > 5.0(sharp peaks)和NSC > 1.5(broad peaks),input样本应具有低S/N,因此NSC值应<2.0。背景均匀度(Bu)。Bu反映背景区域的reads分布偏差,范围从0到1。Bu值低(<0.8)表明reads分布比预期更集中或具有偏好性,通常会导致获得的peaks中出现许多假阳性。对于具有广泛拷贝数变异的基因组(例如MCF-7细胞),则需要宽松的Bu阈值(>0.6)。GC 峰偏差。反映免疫沉淀和PCR扩增过程中的偏好,通常ChIP-seq数据的GC峰值与参考基因组相似。(例如,人类约为50%)。GC偏差(例如,人类超过60%)经常由于PCR扩增偏好和/或来自与CpG岛相关的“超富集”区域的假阳性峰而表现出来。
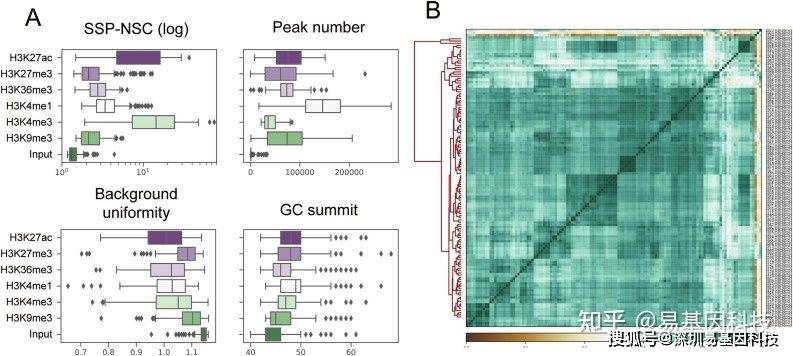
图2:使用 ROADMAP组蛋白修饰数据的QC 分析。(A)六个组蛋白修饰和input样本的四个QC评分分布;(B)Roadmap表观基因组数据库的 117 种细胞类型的 H3K36me3 reads分布的 Pearson 热图。
(7)可视化
在为ChIP-seq数据开发了各种统计方法和质量指标后,reads分布的可视化检查可以有效直观地评估和分析所获得的数据。可以使用交互式可视化工具,如IGV或 SeqMonk。几个web服务器(如UCSC genome browser和WashU Epigenome browser)可以将获得的ChIP-seq结果与其他注释数据关联分析,如进化保守性和各种组织中的基因表达。
(8)归一化比较分析
在比较分析之前,reads归一化对减少技术偏差至关重要。通常使用简单的总reads归一化,将样本reads缩至一致,假设样本之间的比对reads差异充分小于总reads数。但这个假设并不总是得到满足,因此开发了几种方法来鉴定两种条件之间的差异富集区域,其中一些专门为组蛋白修饰数据设计。由于潜在的统计假设不同,不同工具获得的结果差异很大,因此方法的选择将对结果产生关键影响。
两组以上的定量比较更为复杂。当样本之间的预期S/N值相似时,可以使用差异基因表达分析的统计方法。当样品最常见peaks的S/N相似时(如所有样本的单个抗体),也可以利用分位数归一化。如果样本的S/N变化很大(如有和无刺激),则考虑spike-in分析(也称校准分析),该方法在免疫沉淀之前或之后将不同物种的等量DNA添加到所有样本中,并根据衍生reads数估计权重系数。与计算相对差异的归一化方法不同,spike-in ChIP-seq可以研究绝对水平差异。然而即使在归一化后,定量ChIP-seq比较仍然经常受到多步骤样品制备引起的干扰。在这种情况下,可能需要进行简单的两两比较(识别相同或特异性peaks),但所得结果中可能会有一些假阳性和假阴性。
(9)使用DROMPAplus进行ChIP-seq分析
DROMPAplus软件具有许多有价值的功能。首先,它接受各种input map文件格式,包括CRAM。其次,DROMPAplus可以在一行中显示两个样品(图3A),描绘了reads富集的共现关系(如H3K4me3和H3K27ac)和特异性(如H3K27me3和H3K36me3),染色质环从3C鉴定中获得。第三,使用SSP从单端reads中自动计算片段长度。第四,该软件可高度定制轨道高度(track heights)、坐标轴范围(axis limits)和显示功能(display features)。例如该软件可以通过用linear-scale和log-scale突出显示富集区域来描述ChIP/input富集。第五,DROMPAplus可以支持spike-in归一化和总reads归一化。最后,它使用比C语言更灵活的C++语言,计算速度比Python和R快。
图3:使用DROMPAplus可视化多个ChIP-seq样本。
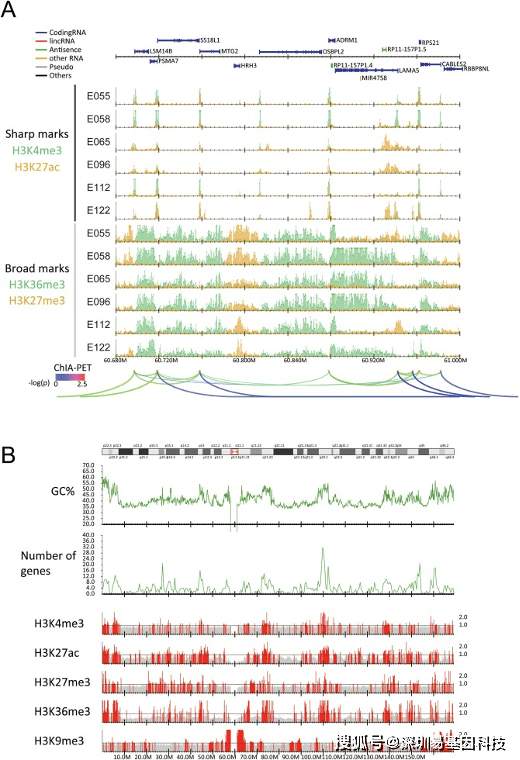
(A) E055(包皮成纤维细胞),E058(包皮角质形成细胞),E065(主动脉),E096(肺),E112(胸腺)和E122(人脐静脉内皮细胞:HUVEC)的sharp(上)和broad(下)两个组蛋白标记的归一化reads数分布。HUVEC中RNA Pol II介导的染色质环(基于ChIA-PET数据)由arches表示。
(B)人7号染色体的五个主要组蛋白修饰(E122)的可视化。
(10)功能分析
Motif分析研究了peaks或特定表观基因组区域(如增强子位点)中的特异性序列,并预测鉴定区域内可能的转录因子结合位点。一般来说,motif分析方法可以分为两种类型:
- de novo motif discovery,用于鉴定出现在大部分peaks中未知因子的潜在新结合motif;
② motif scanning,用于预测和排列提供的DNA序列与数据库中所有已知motif的相似性。
ChIP-seq peaks也可用于功能富集分析。该分析将附近基因作为潜在靶点进行双向标记或定量排序,并通过GO或KEGG分析对其进行分组。
(11)染色质状态注释
染色质状态注释,也称为半自动基因组注释(semi-automated genomic annotation,SAGA),使用非监督学习方法,通过特异性表观基因组模式(如启动子,增强子,转录区域和抑制区域)对所有基因组区域进行分类。获得的簇(clusters)被注释为染色质状态,特异性区域分析(如增强子分析)缩小了待研究的目标基因组区域。而染色质状态注释使用隐马尔可夫模型(Hidden Markov Model,HMM)或动态贝叶斯网络(Dynamic Bayesian Network, DBN)分割基因组并将染色质状态分配给全基因组区域。在此分析中,生物学上最佳的状态数量必须通过实验定义。
获得的染色质状态进一步扩展用于各种下游分析。如ChromDiff、EpiCompare和ChromDet将多种细胞类型的表观基因组景观组合并聚类,以研究组织或细胞类型特异性表观基因组区域。还可利用概率聚类方法来捕获多个细胞系和多个时间点的染色质动态变化。基于图的正则化(Graph-based regularization ,GBR)整合染色质互作信息,用于染色质状态注释。生成的染色质状态信息用于揭示个体遗传变异和了解进化中的表观遗传变异。
三、ChIP-seq高级应用(Advanced applications)
研究中常用的的细胞类型具有十分丰富的ChIP-seq数据,利用这些细胞类型的信息可以推断基因组动力学或通过较少的相关实验可以注释其他细胞类型的表观遗传景观。越来越多证据表明,表观遗传信息与基因表达和染色体构象高度相关,且可预测。以下简要介绍用于组蛋白修饰的ChIP-seq分析的高级应用工具。
(1)通过表观基因组信息预测基因表达
目前已经开发了对ChIP-seq实验获得的表观遗传信息定量预测基因表达水平的各种基于机器学习方法。如Karlic等人将线性回归模型(linear regression model)应用于启动子位点的组蛋白修饰富集,以预测CD4+ T细胞基因表达;他们利用19种组蛋白修饰,并提出只要3个启动子位点修饰就足以模拟基因表达。Dong等人使用非线性模型(non-linear models),如多元自适应样条回归(multivariate adaptive regression splines ,MARS)和随机森林(random forests),在7种人细胞系中绘制了11种组蛋白修饰和DNase I超敏反应,并成功预测了基因表达水平(Pearson系数r = 0.83)。以上模型仅考虑启动子位点的表观遗传模式,而不考虑增强子位点信息。
DeepExpression利用HiChIP数据,一种用于捕获以蛋白质为中心的染色体环高通量技术,用于探索增强子和增强子-启动子互作。此外,还可以使用卷积神经网络(convolutional neural networks ,CNN)来预测基因表达或差异基因调控模式。将多个组蛋白修饰ChIP-seq数据和HiChIP数据相比,由于制备单个RNA-seq样品需要相对较低的成本,研究主要目的是阐明组蛋白修饰在基因调控中的组合作用,而不是预测基因表达水平本身。
(2)从表观基因组数据预测染色质互作
已有证据表明增强子中的单核苷酸多态性(SNPs)可以引起遗传性疾病和癌症,因此非常需要全基因组分析来表征增强子在特定细胞系中的作用。然而,增强子和靶基因的全基因组配对并非易事。增强子不一定调节最近的基因,甚至一些增强子远离TSS。尽管染色体构象捕获(3C)检测(如Hi-C,HiChIP和ChIA-PET),可对整个基因组的空间接近度进行定量分析,但用于配对增强子和靶标的计算工具在不断发展。Hariprakash和Ferrari将基因增强子配对工具分为四类:基于相关性,基于监督学习,基于回归和基于评分。主要区别在于“是否每个基因考虑多个增强子”和“是否每个增强子/启动子位点考虑多个表观遗传数据”。基于相关性方法分析所有增强子-启动子互作关系,而基于回归方法假设多个增强子可作用于单个基因。基于监督学习和基于评分的方法可以为每个位点组合多个ChIP-seq数据集和其他信息类型(如进化保护)。以上工具专注于增强子-启动子互作,同时,还有许多其他染色质互作,如增强子-增强子环和相分离的弱染色质耦合。CITD和DRAGON分别使用小波变换(wavelet transformation)和势能函数(potential energy functions)从表观遗传数据中全面解析三维基因组组织。
以上统计方法旨在找到与空间染色质接触相关表观遗传数据中的一致模式,并在没有任何基因组架构知识的情况下进行预测。其局限性在于基因组互作是定性而并非定量,尽管具有动态属性。在training和验证数据集中共享基因组结构信息,目前的统计方法可能存在training偏差。由于工具的数量正在快速增长,未来的方法可能会达到足够的准确性,从而不需要通过3C数据来鉴定增强子-启动子互作。
(3)数据归集(data imputation): ChIP-seq数据重建和去噪
大规模ChIP-seq分析中的一个分析挑战来自ChIP-seq数据中的偏差和批次效应。机器学习方法对training数据噪敏,不可避免地将一些ChIP-seq样本判定为中等质量或低质量数据(导致数据丢失),尤其在多个实验室负责的数据采集情况下(如大型联合项目)。如果样品非常珍贵(如原代细胞和临床样品),实际上可能难以收集更多样本。这种情况下,“data imputation”方法可能可以利用其他密切相关细胞类型的表观遗传数据进行数据去噪或重建。
“数据去噪(Data de-noising)”旨在通过鉴定和去除数据中的噪声来改善现有的ChIP-seq数据质量。例如Coda对生成噪声过程进行编码,并使用卷积神经网络(CNN)恢复ChIP-seq数据中的信号。“数据重建(Data reconstruction)”旨在从silico的大型数据集中生成缺失的ChIP-seq数据。ChromImpute是一种开创性的工具,可以使用十种最相关细胞类型训练回归树(regression tree),从每个缺失的实验推断信号。PREDICTD和Avocado利用张量分解(tensor decomposition)同时分析多个ChIP-seq数据。
data imputation是ChIP-seq实验的潜在替代方法,并且可能为收集生物学中所有可能细胞类型和环境条件的表观基因组数据开辟道路。在现阶段,预测与其他样本不相关的样本特异性信号与遗传变异的结合存在局限性。即使在高质量数据可用的情况下,通过基因组数据归集的“信号先验期望(a prior expectation of signal)”也能提供信息,所以检测数据和测算数据的结合使用是一种较好策略。
四、单细胞 ChIP-seq 分析
已有证据表明,许多细胞类型(包括正常免疫细胞)在复杂的组织和肿瘤中起着重要的辅助作用。为阐明发育过程中的细胞异质性和细胞命运轨迹(cell fate trajectories),研究人员开发了各种单细胞分析方法。其中scChIP-seq能够在低输入(low-input)样品情况下以单细胞分辨率对组蛋白修饰和其他染色质结合蛋白进行全基因组分析。下表为利用微流控系统(microfluidic systems)、Tn5转座酶标记(Tn5 transposase tagmentation)和ChIP-free策略的多种用于单细胞标记(tagmentation)和ChIP-seq文库制备方法(表2)。
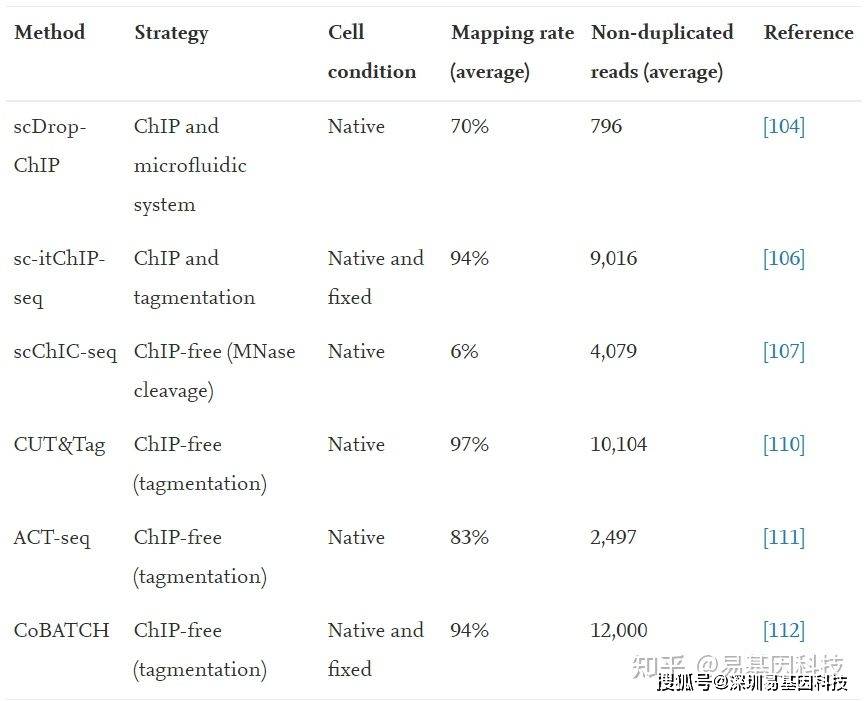 表2:scChIP-seq方法
表2:scChIP-seq方法
(1)基于微流控系统的方法
第一种scChIP-seq方法scDrop ChIP使用微流体系统进行细胞标记,并结合常规ChIP方法生成在每个细胞中产生约800个不重复(non-duplicated)reads。液滴微流控(droplet microfluidic)技术提供了更高的分辨率,每个细胞产生约10000个不重复reads。这些方法的局限性在于大多数实验室通常不使用专用的微流控装置。
(2)基于标记的方法
使用Tn5转座酶标记的文库制备已广泛用于包括ChIP-seq在内的各种NGS分析。sc itChIP-seq在常规ChIP实验之前采用标签化技术进行单细胞标记和文库制备,可以在每个细胞中产生约9000个不重复reads。由于该方法的实验过程与常规ChIP-seq方法相似,因此比scDrop-ChIP更易于使用。
(3)ChIP-free方法
scChIP-seq开发了几种ChIP-free方法:单细胞染色质免疫裂解测序(scChIC-seq)和单细胞uliCUT&RUN,它们基于CUT&RUN方法,采用MNase和蛋白A融合蛋白(protein A fusion proteins)检测具有特异性抗体的裂解靶点。通过严格实验步骤进行文库制备,每个细胞可产生约4100个不重复reads,缺点是reads比对率比较低(∼6%)。另外还有三种类似的方法:CUT&Tag、ACT-seq和CoBATCH,这几种方法使用Tn5转座酶和蛋白a融合蛋白检测。在文库制备过程中,目标蛋白结合在染色体上后,融合蛋白捕获一级抗体(primary antibody);然后激活Tn5转座酶并在蛋白质结合位点进行标记。这几种方法的优点是可以同时进行蛋白质结合位点检测和文库制备,大大减少了实验步骤和时间,且较少受到免疫沉淀步骤偏差的影响,每个细胞可显示约 97%的比对率并产生约12000个不重复reads。因此,这种ChIP-free方法具有高通量和高质量scChIP-seq分析的潜力。
最后,染色质整合标记测序(Chromatin Integration Labeling sequencing ,ChIL-seq)是一种基于免疫染色的ChIP-free方法。该方法使用与dsDNA偶联的二抗探针(secondary antibody probe),该探针包含T7 RNA聚合酶启动子、NGS接头序列和Tn5结合序列,在捕获第一抗体后,通过Tn5转座酶将探针DNA序列整合到目标结合位点。然后通过转录扩增整合区域,进行RNA纯化和文库制备。该方法可用于单细胞分析,但可能需要进行多次优化才能实现高通量测序。
在未来,将会开发更多scChIP-seq方法,如同时检测多个组蛋白修饰和其他染色质结合蛋白。这些研究将能够捕获每个细胞染色体上的基因调节因子的互作关系。
五、总结
这篇综述主要讨论了组蛋白修饰ChIP-seq分析的概念和方法。作者阐明了从质量评估到染色质状态注释标准分析的逐步工作流程,重点阐述与每个步骤相关的关键点。然后讨论了几种使用机器学习方法的高级ChIP-seq应用。由于表观基因组数据的可用性越来越高,使用这些数据鉴定新基因组特征的工具将继续受到关注。
“高级应用”介绍的几乎所有方法都使用有监督机器学习方法(如deep-CNN),其中一个局限性在于这些方法需要每个细胞系的许多样品来开发training数据,从而导致需要大量的ChIP-seq数据。此外,这些方法需要在分析环境中具有强大的计算能力和足够的磁盘存储,云计算可能是克服此限制的一种解决方案。研究人员可以通过云计算共享PB数据集和计算环境,大大降低了大规模重新分析公共数据的计算成本。另一个局限是input数据的准确性,初始样品的training数据通常是从各种NGS分析中获得,其中包含了技术/生物噪声,对有效training有很大的干扰。尽管data imputation方法在一定程度上突破了这种限制,但仍有必要为training数据开发经过实验验证的“黄金标准数据集(gold standard datasets)”以评估工具性能。
此外ChIP-seq分析的另一个挑战是与其他NGS测序的联合分析(如DNA甲基化、开放区域(accessible regions)和3D基因组折叠(3D genome folding))。其中Hi-C等三维基因组信息可以预测与表观遗传特征密切相关的增强子-启动子互作和拓扑结构域(Topologically associating domains,TAD)。最后作者讨论了近期开发的scChIP-seq分析方法,多个scChIP-seq方案促进了相关算法的发展。
关于染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
- 转录因子和辅因子结合作用复制因子和 DNA 修复蛋白组蛋白修饰和变异组蛋白
技术优势:
- 物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:
参考文献:
Ryuichiro Nakato, Toyonori Sakata,Methods for ChIP-seq analysis: A practical workflow and advanced applications,Methods,Volume 187,2021,Pages 44-53,ISSN 1046-2023,https://doi.org/10.1016/j.ymeth.2020.03.005.
http://www.egenetech.com- 本文固定链接: https://maimengkong.com/zu/1442.html
- 转载请注明: : 萌小白 2023年4月15日 于 卖萌控的博客 发表
- 百度已收录
