2014年,单细胞测序被列为《Nature Methods》年度最重要的方法学进展。通常说的单细胞测序,是指单细胞的mRNA测序。随着单细胞RNA测序技术日渐成熟,可预计将来会有越来越多的单细胞测序应用于科研和临床。细心的读者可能发现,转录时空已经推送了很多单细胞测序领域的文章。针对火热的单细胞测序,我们将对单细胞测序的特点、实验设计、分析方法将一一进行介绍,分享给大家。
1-RNA测序特点
a) “大体积”(Bulk)RNA-seq
2000年以来的主要技术突破(替代microarray),现已广泛使用
以大量细胞来检测基因的平均表达水平
可应用于比较转录组学,如不同物种的同种组织样本
可应用于基因定量
不适合用于研究具有异质性的系统,如早期发育,复杂组织(脑)
不能表征基因表达的随机性( stochastic)本质
b) 单细胞RNA测序
2009年汤富酬等人年首次报道的新技术
直到约2014年,由于新的单细胞测序方法提出和测序成本的大幅下降,单细胞测序才开始广泛使用
可定量单细胞基因表达水平,并可获得一群细胞的基因表达水平分布
对于研究转录组在单细胞水平特异性变化等问题非常有用,如细胞类型鉴定、细胞应答异质性、基因表达随机性、细胞间的基因调控网络
大多数情况下,现有的数据分析方法需要调整要才能使用,或者开发新的分析方法
不同的单细胞测序建库方法有:SMART-seq2(2013), CEL-seq(2012), SCRB-seq(2014), Drop-seq(2015)
2-单细胞测序实验流程
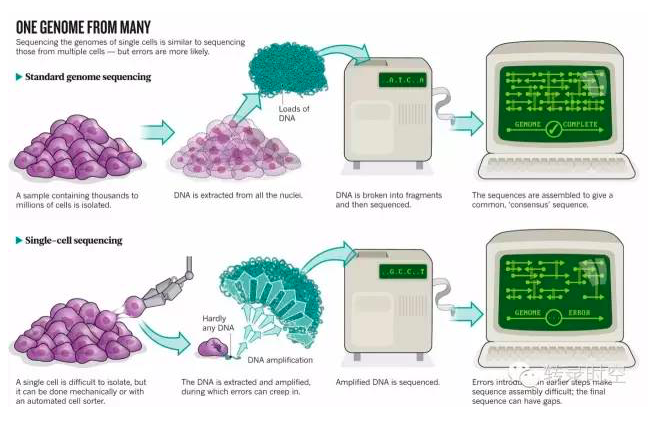
总体来说,单细胞测序的方法与大体积RNA测序方法类似,对于具体实验流程的讨论,本文不在此赘述。总结如下
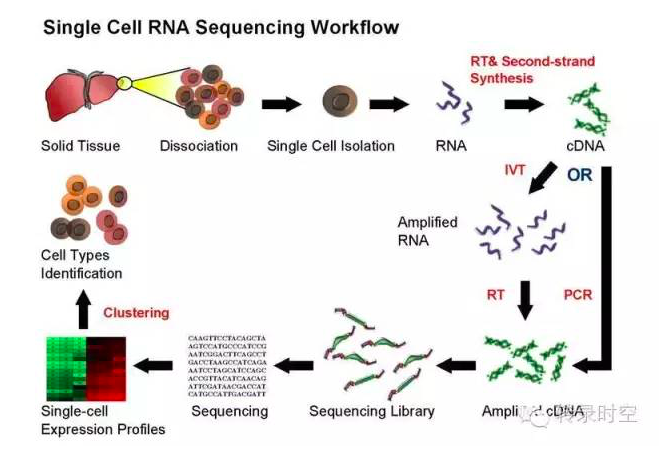
3-单细胞测序数据分析流程
第一步(黄色):跟一般高通量测序一样,检查测序序列质量,序列回帖,检查序列比对质量;
第二步(橘黄色):需要结合现有的RNA-seq分析方法和新的方法来处理单细胞测序技术上的不同。最大的不同点在于normalization;
最后:对单细胞RNA-seq的数据解读应使用特为单细胞开发的分析方法。
我们将在下期详细介绍以下各步骤涉及到的分析工具和方法
4-单细胞测序的挑战
与“大体积”RNA-seq不同,单细胞RNA-seq的主要区别在于一个测序文库代表一个细胞,而不是大量的细胞(Drop-seq例外,其利用细胞UMI和分子UMI将大量细胞建成一个文库)。因此,须加倍关注单细胞测序文库之间的不同。文库间的差异性主要体现在:
扩增效率(Amplification),差异可达100,000倍
基因“丢失”(dropouts),即一些基因在某些细胞中有表达,但是在另一些细胞中无法检测到
这两种差异都是由于起始材料太过稀少(RNA分子从一个细胞而来)而产生的。提高转录本捕获效率和降低PCR扩增偏好性是亟待解决的问题, 因此这个领域的研究也相当活跃。
5-单细胞测序的对照
为了更好估计和消除单细胞测序文库间的技术(系统)误差,现有两种定量标准被广泛采用,即spike-ins和UMIs。使用这两种对照是为了辅助规范(normalization)不同细胞间的基因表达水平。
Spike-ins
Spike-ins是已知浓度的外源RNA分子。在单细胞裂解液中加入Spke-ins后,再进行反转录。最广泛使用的Spike-ins是External RNA Control Consortium (ERCC)提供的合成spikes。其包含96个不同长度和GC含量的mRNA分子 (Jiang et al. 2011)。但是spike-ins的使用浓度通常很高,结果会占据很大比例的测序reads。最新的Drop-seq技术也还没不能加入spike-ins。
UMIs
另一种标准化方法是使用 Unique Molecular Identifiers (UMIs)(Kivioja et al. 2012). UMIs是一种随机条形码(barcode)序列,长度在4-20 bp之间。 在扩增步骤之前(通常在反转录期间),UMIs被添加在每个转录本cDNA的3’或5’端。之后,对转录本末端进行靶向测序。这些barcodes使得在扩增步骤之前,可以对转录本进行定量。虽然UMIs消除扩增偏好性的效果非常好,但是不合适用于研究基因异构体和allel特异表达。
更多内容,下回分解
参考文献:
Tang, Fuchou, Catalin Barbacioru, Yangzhou Wang, Ellen Nordman, Clarence Lee, Nanlan Xu, Xiaohui Wang, et al. 2009. “mRNA-Seq Whole-Transc riptome Analysis of a Single Cell.” Nat. Methods 6 (5): 377–82.
Picelli, Simone, Åsa K Björklund, Omid R Faridani, Sven Sagasser, Gösta Winberg, and Rickard Sandberg. 2013. “Smart-Seq2 for Sensitive Full-Length Transc riptome Profiling in Single Cells.” Nat. Methods 10 (11): 1096–8.
Hashimshony, Tamar, Florian Wagner, Noa Sher, and Itai Yanai. 2012. “CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification.” Cell Rep. 2 (3): 666–73.
Macosko, Evan Z, Anindita Basu, Rahul Satija, James Nemesh, Karthik Shekhar, Melissa Goldman, Itay Tirosh, et al. 2015. “Highly Parallel Genome-Wide ex pression Profiling of Individual Cells Using Nanoliter Droplets.” Cell 161 (5): 1202–14.
Stegle, Oliver, Sarah A Teichmann, and John C Marioni. 2015. “Computational and Analytical Challenges in Single-Cell Transc riptomics.” Nat. Rev. Genet. 16 (3): 133–45.
Jiang, Lichun, Felix Schlesinger, Carrie A Davis, Yu Zhang, Renhua Li, Marc Salit, Thomas R Gingeras, and Brian Oliver. 2011. “Synthetic Spike-in Standards for RNA-seq Experiments.” Genome Res. 21 (9): 1543–51.
Kivioja, Teemu, Anna Vähärautio, Kasper Karlsson, Martin Bonke, Martin Enge, Sten Linnarsson, and Jussi Taipale. 2012. “Counting Absolute Numbers of Molecules Using Unique Molecular Identifiers.” Nat. Methods 9 (1): 72–74.- 本文固定链接: https://maimengkong.com/kyjc/714.html
- 转载请注明: : 萌小白 2021年7月16日 于 卖萌控的博客 发表
- 百度已收录
