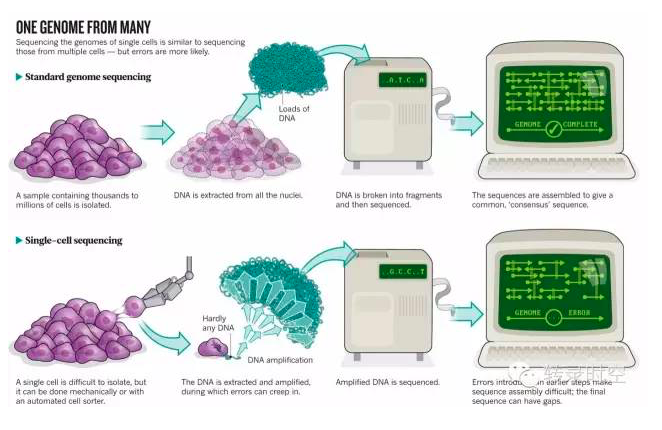
尽管从一群细胞中获得全基因组范围的mRNA表达量对理解生物学机制非常有用,但这种方法只能获得平均基因表达水平,有时候会掩盖,甚至误导一些有意思的生物学现象。幸运的是,随着高通量测序技术的迅速发展,我们可以获得单个细胞的转录组。
与大体积(bulk)RNA测序相比,单细胞RNA测序的数据结构虽然与其相同,但也有它不一样的特征:起始RNA极其稀少、分辨率高(resolution,即存在大量的基因有0值表达的情况,图1a)、异质性高(variability,图1b)、基因表达的分布复杂(distribution,图1c)[1]。
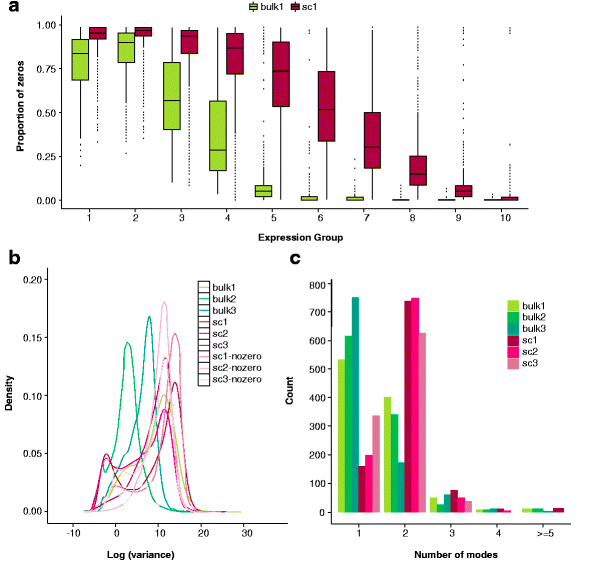
图1:单细胞与大体积RNA-seq的异同
sc:单细胞测序,bulk:大体积测序
针对单细胞测序的特点,我们先讨论单细胞实验设计、质量评估和表达定量方面应该注意的问题。
1-单细胞测序的实验设计
为了避免有意思的生物学现象才不被样本准备或数据收集过程中产生的假象和额外混杂变量(confounding)所掩盖,应把各种相关变量随机化处理。需要注意的是,在实验过程中,常常因为资源有限(如样本、单细胞分离设备、测序仪、预算等),完全随机化的设计并不现实。大多数情况下,样本是分批次(batch)准备的。例如分离胚胎发育过程中的1, 2, 4 , 8期的细胞:每个批次细胞应包含各期不同发育阶段的细胞。这样可使批次效应与发育时段变量不会混杂(confound)。Hicks等人[2]提出一种合理的实验设计为:在每个批次的样本中应同时出现不同生物学处理的样本,测序过程应根据大体积测序方法随机化样本处理(sequencing run,flow cell, lanes等)。基于此设计,数据分析人员易从系统实验偏好(bias)中调整批次效应等无关变量。
单细胞测序实验的建库方法、测序平台和单细胞分离方法都会对测序覆盖度(coverage)和链特异性产生一定的影响。另外,是否要加入spike-ins,以及UMIs也是需要慎重考虑的问题。Spike-ins的使用浓度通常很高,结果会占据很大比例的测序reads。最新的Drop-seq技术也还不能使用spike-ins。UMIs在消除扩增偏好性方面非常有用,但是不能用于研究基因异构体和allel特异表达。
总而言之,记录越多的影响因素,越对下游数据分析有利。另外,影响单细胞测序的变量也很可能来源于系统误差。
2-单细胞测序的质量评估
在计算各个细胞的表达量之前,应对原始测序数据、转录组比对数据、单细胞收集质量进行评估,并去除低质量细胞。低质量是指:细胞破裂或死亡、捕获单细胞的设备上没有细胞、设备上有不止一个细胞存在[3]。直接用显微镜镜检的方法可以去掉低质量样本。大体积测序常用的质量评估软件如FastQC、Kraken、RSeQC也可以用于检查测序后的细胞质量。
如使用FastQC:
$fastqc experiment.fq
转录组比对数据的检查也是常用的质量评估手段。获知有多少测序序列比对到rRNA/tRNAs、特异比对到基因组的测序序列、跨外显子的测序序列和转录本的测序深度等信息非常有助于细胞质量的评估。
如使用RSeQC:
$python/geneBody_coverage.py -i input.bam -r genome.bed -o output.txt
$python/bam_stat.py -i input.bam -r genome.bed -o output.txt
$python/split_bam.py -i input.bam -r rRNAmask.bed -o output.tx
需要注意的是,质量评估的结果应参考单细胞测序的实验方案。选择带polyA的RNA进行测序(去除rRNA),其测序的覆盖度会产生3’偏好性。如下图所示,3个有严重3’偏好性的细胞应在下游定量分析中去除。
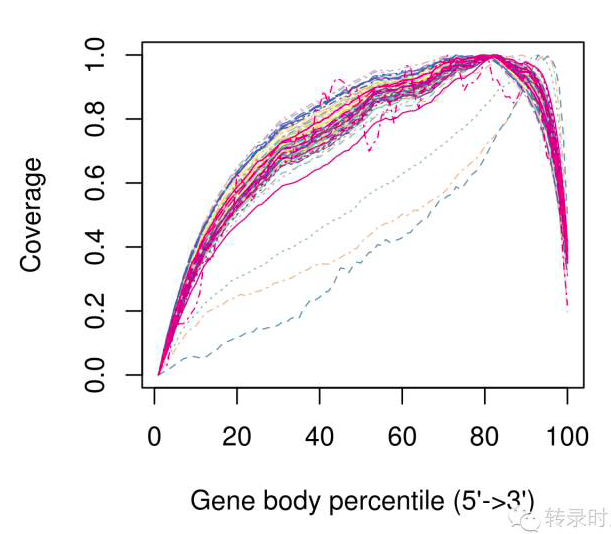
3-单细胞测序的表达定量
下一步就是对每个细胞基因表达水平进行定量。对于mRNA来说,可以直接使用大体积测序的定量工具,常用工具有:HTseq、FeatureCounts、RSEM和WemIO等。
如使用FeatureCounts:
# include multimapping
$featureCounts -O -M -Q 30 -p -a genome.gtf -o outputfile input.bam
# exclude multimapping
$featureCounts -Q 30 -p -a genome.gtf -o outputfile input.bam
值得一提的是,大体积测序中用于表征相对表达量的方法(FPKM/RPKM,TPM)不适用于单细胞测序。这些方法假设每个细胞的RNA总量相同,且相同基因在不同细胞中表达量一致。因基因表达具有随机性( stochastic),该假设不成立。因此,单细胞测序实验中最常使用UMIs,以获知单细胞内mRNA分子的绝对数量,然后对不同细胞的基因表达量进行比较。- 本文固定链接: https://maimengkong.com/kyjc/715.html
- 转载请注明: : 萌小白 2021年7月16日 于 卖萌控的博客 发表
- 百度已收录
