转录组测序的最直接目的,就是设法寻找组间显著表达变化的基因,解释基因表达水平的变化对生物功能的影响。例如,肿瘤患者与正常人群相比哪些编码蛋白或非编码RNA分子发生了失调,这些失调分子是否是引发或加速肿瘤进程的潜在因素?逆境胁迫下的植物体中哪些基因表达显著激活,这些激活的基因是否有利于植物应对高温、干旱、盐胁迫等不利环境。
那么,如何基于转录组测序获得的定量表达值,识别差异表达变化的基因或其它非编码RNA分子呢?
查阅相关文献寻找思路的习惯是一定要养成的,文献的材料方法或者正文部分都会有相关的描述,使用了哪个工具(例如DESeq2)作差异表达分析,阈值是什么(例如倍数变化≥2,p<0.05等)。本篇就以R包DESeq2的差异表达基因分析方法为例做个简单演示,这是目前使用频率最高的鉴定差异分子的R包之一。
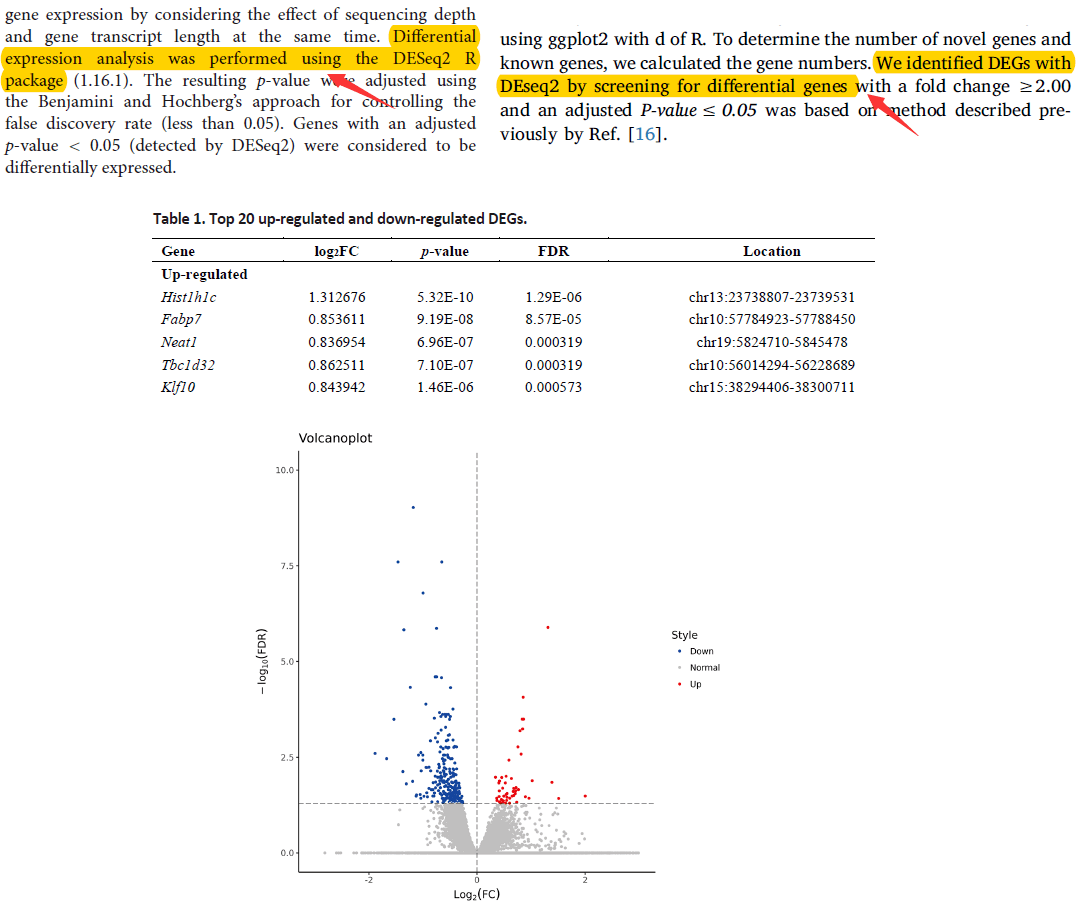 相关文献描述
相关文献描述
1 关于DESeq2包的安装
对于DESeq2的安装也很简单,一般情况下,直接通过Bioconductor安装DESeq2就可以了。
考虑到19年下半年的时候,DESeq2做过重大更新和优化,核心算法没变,但是运行速率相对于先前的版本提升了几十倍!小编不清楚最新版本的DESeq2是否已经添加至Bioconductor中,因此如果想安装最新版本提高计算效率,而又不确定Bioconductor的DESeq2是否是新版本的情况下,建议直接链接至作者的github库中直接安装。
!!!**************************************************
##以下两种安装 DESeq2 的方法二选一
#(1)常规方法 bioconductor 安装
#install.packages('BiocManager') #需要首先安装 BiocManager,如果尚未安装请先执行该步
BiocManager::install('DESeq2')
#(2)在 GitHub 中获取最新版本的方式
#install.packages('devtools') #需要首先安装 devtools,如果尚未安装请先执行该步
devtools::install_github('mikelove/DESeq2@ae7c6bd')
#其它备注项:若中间提示有其它依赖 R 包的旧版包冲突的话,先删除旧包再安装新的
remove.packages('xxx')
BiocManager::install('xxx')
!!!**************************************************
2 DESeq2分析差异表达基因的一般过程
DESeq2是一种基于负二项式分布的方法,使用局部回归推算均值和方差,通过离散度和倍数变化的收缩率估计以提高稳定性。定量分析关注的更多是差异表达的“强度”,而非“存在”。
以下是DESeq2分析差异表达基因的一般过程。
2.1 准备数据和文件读取
首先准备基因表达值矩阵。
本文的所有测试数据和R代码,可在文末获取
- “control_treat.count.txt”,是6个测序样本的基因表达值矩阵,包括3个处理组(treat)和3个对照组(control)。第1列是基因名称,注意不能有重复值。
 示例文件样式,行是基因,列是样本,内容为基因表达值
示例文件样式,行是基因,列是样本,内容为基因表达值
然后将准备好的基因表达值矩阵读到R中,并预定义样本分组信息。
!!!**************************************************
#读取基因表达矩阵
dat <- read.delim('control_treat.count.txt', row.names = 1, sep = '\t', check.names = FALSE)
#指定分组因子顺序
#注意要保证表达矩阵中的样本顺序和这里的分组顺序是一一对应的
coldata <- data.frame(condition = factor(rep(c('control', 'treat'), each = 3), levels = c('control', 'treat')))
!!!**************************************************
2.2 计算差异倍数列表
整体过程非常简单,因为作者已经将多步过程整合到一个函数中,因此使用起来非常的方便。大致上,DESeq2两步就完成分析了。
- 第1步是构建DESeqDataSet对象,包括对表达值的标准化以及存储输入值和中间结果等。
- 第2步,函数DESeq()是一个包含因子大小估计、离散度估计、负二项模型拟合、Wald统计等多步在内的过程,结果将返回至DESeqDataSet对象。最后获得各基因的差异倍数变化和显著性p值,用于后续的差异基因筛选。
!!!**************************************************
##DESeq2 默认流程
library(DESeq2)
#第一步,构建 DESeqDataSet 对象
dds <- DESeqDataSetFromMatrix(countData = dat, colData = coldata, design= ~condition)
#第二步,计算差异倍数并获得 p 值
#备注:parallel = TRUE 可以多线程运行,在数据量较大时建议开启
dds1 <- DESeq(dds, fitType = 'mean', minReplicatesForReplace = 7, parallel = FALSE)
#注意,需将 treat 在前,control 在后,意为 treat 相较于 control 中哪些基因上调/下调
res <- results(dds1, contrast = c('condition', 'treat', 'control'))
res
!!!**************************************************
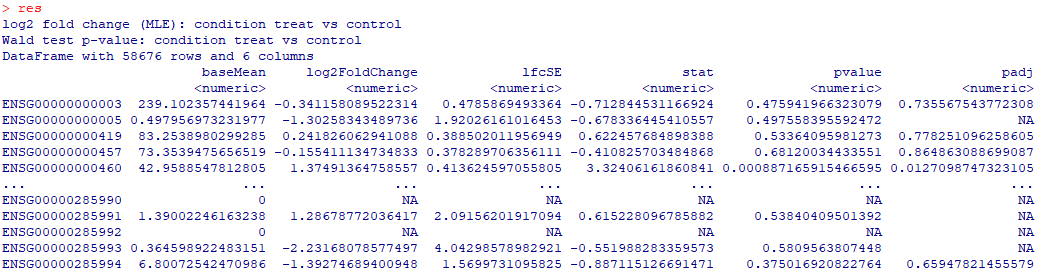 DESeq2默认输出
DESeq2默认输出
差异分析结果保存在“res”中,包含了基因id、标准化后的基因表达值、log2转化后的差异倍数(Fold Change)值、显著性p值以及校正后p值(默认FDR校正)等主要信息。
如果期望将该表格输出到本地,转化为数据框结构后直接write.table()就可以了。
!!!**************************************************
#输出表格至本地
res1 <- data.frame(res, stringsAsFactors = FALSE, check.names = FALSE)
write.table(res1, 'control_treat.DESeq2.txt', col.names = NA, sep = '\t', quote = FALSE)
!!!**************************************************
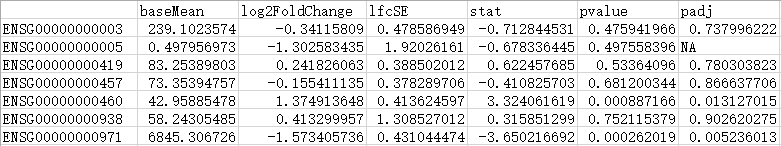 DESeq2结果的本地输出
DESeq2结果的本地输出
2.3 筛选差异表达基因
后续通过该表,即可自定义阈值筛选差异表达基因了。
- 例如这里期望根据|log2FC|≥1以及padj<0.01筛选,并通过“up”和“down”分别区分上、下调的基因。
!!!**************************************************
##筛选差异表达基因
#首先对表格排个序,按 padj 值升序排序,相同 padj 值下继续按 log2FC 降序排序
res1 <- res1[order(res1$padj, res1$log2FoldChange, decreasing = c(FALSE, TRUE)), ]
#log2FC≥1 & padj<0.01 标识 up,代表显著上调的基因
#log2FC≤-1 & padj<0.01 标识 down,代表显著下调的基因
#其余标识 none,代表非差异的基因
res1[which(res1$log2FoldChange >= 1 & res1$padj < 0.01),'sig'] <- 'up'
res1[which(res1$log2FoldChange <= -1 & res1$padj < 0.01),'sig'] <- 'down'
res1[which(abs(res1$log2FoldChange) <= 1 | res1$padj >= 0.01),'sig'] <- 'none'
#输出选择的差异基因总表
res1_select <- subset(res1, sig %in% c('up', 'down'))
write.table(res1_select, file = 'control_treat.DESeq2.select.txt', sep = '\t', col.names = NA, quote = FALSE)
#根据 up 和 down 分开输出
res1_up <- subset(res1, sig == 'up')
res1_down <- subset(res1, sig == 'down')
write.table(res1_up, file = 'control_treat.DESeq2.up.txt', sep = '\t', col.names = NA, quote = FALSE)
write.table(res1_down, file = 'control_treat.DESeq2.down.txt', sep = '\t', col.names = NA, quote = FALSE)
!!!**************************************************
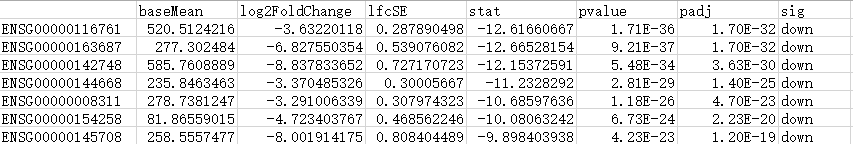 筛选的显著上下调基因
筛选的显著上下调基因
2.4 差异表达火山图示例
已经识别了显著差异表达的基因,最后期望通过火山图将它们表示出来,火山图是文献中常见统计图表之一。
- ggplot2为此提供了优秀的作图方案,参考以下示例。
!!!**************************************************
##ggplot2 差异火山图
library(ggplot2)
#默认情况下,横轴展示 log2FoldChange,纵轴展示 -log10 转化后的 padj
p <- ggplot(data = res1, aes(x = log2FoldChange, y = -log10(padj), color = sig)) +
geom_point(size = 1) + #绘制散点图
scale_color_manual(values = c('red', 'gray', 'green'), limits = c('up', 'none', 'down')) + #自定义点的颜色
labs(x = 'log2 Fold Change', y = '-log10 adjust p-value', title = 'control vs treat', color = '') + #坐标轴标题
theme(plot.title = element_text(hjust = 0.5, size = 14), panel.grid = element_blank(), #背景色、网格线、图例等主题修改
panel.background = element_rect(color = 'black', fill = 'transparent'),
legend.key = element_rect(fill = 'transparent')) +
geom_vline(xintercept = c(-1, 1), lty = 3, color = 'black') + #添加阈值线
geom_hline(yintercept = 2, lty = 3, color = 'black') +
xlim(-12, 12) + ylim(0, 35) #定义刻度边界
p
!!!**************************************************
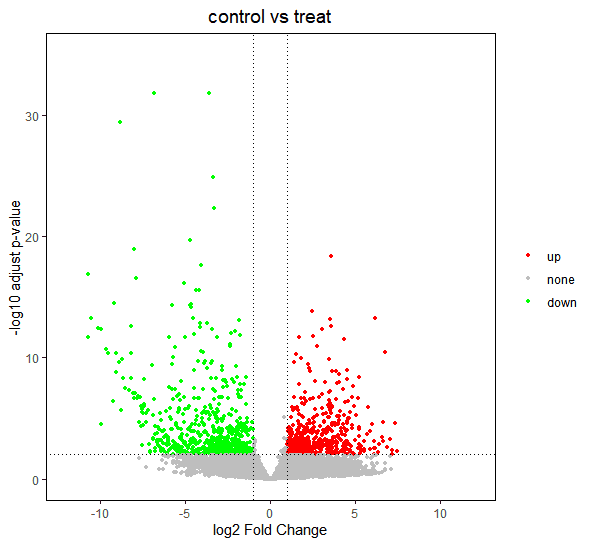 差异表达火山图示例
差异表达火山图示例
注:!!!*******之间为R脚本内容
- 本文固定链接: https://maimengkong.com/zu/878.html
- 转载请注明: : 萌小白 2022年4月29日 于 卖萌控的博客 发表
- 百度已收录
