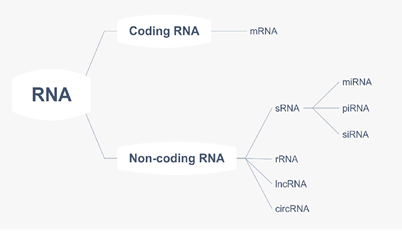
全转录组是指特定组织或细胞在特定状态下转录出的所有转录本信息的总和。与常规转录组相⽐,通过构建链特异转录组⽂库和⼩RNA⽂库的全转录组测序可以同时获得mRNA、lncRNA、circRNA和miRNA四种类型的RNA信息,对其进⾏种类鉴定、定量、差异分析和RNA互作整 合分析。⽬前单⼀的mRNA或ncRNA研究可能⽆法满⾜科研需求,通过全转录组测序和联合多种RNA的整合分析可以更全⾯的解析转录调控机制。
2021年联川生物从心初发,为大家带来了大幅度的更新,增加了circos图加强版、桑吉图、Cibersort免疫浸润反卷积分析、雷达图、UpSet图、STEM时间趋势分析图、四象限图等图表,让你的分析不再枯燥。
当然部分分析物种仅限于人和小鼠,其他非模式物种部分分析仍然无法进行,敬请谅解。
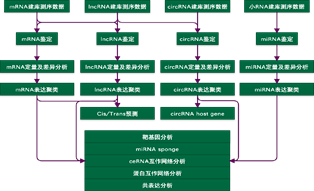
对于全转录组测序而言,通过2个文库可以拿到4套不同RNA的分析结果,以miRNA为核心可以进行多组学关联分析。
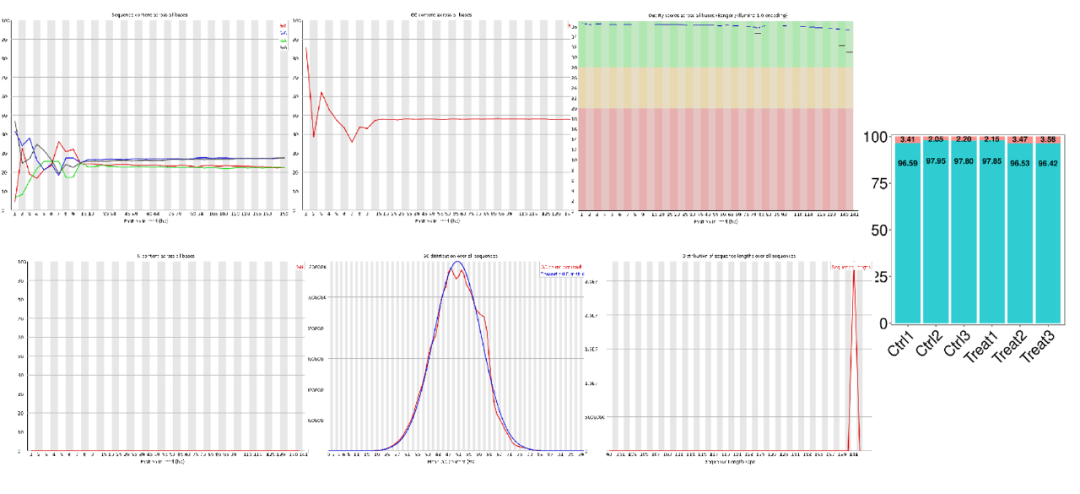
原始数据以fastq⽂件格式存储,在fastq格式⽂件中每个reads有四⾏描述,包含碱基序列信息和质量值,质量值⽤ASCII码表示。碱基的测序 错误率Perror和质量值的关系为Q = -10log10Perror;即Q20和Q30对应测序的错误率分别为1%和0.1%。

⼀般转录组的序列⽐对可以以基因组或转录组(⽐如三代全⻓转录组)作为参考。以转录组为参考的分析,⽐较适合基因注释⽐较完善的 物种(⽐如⼈、⼩⿏),同时⽐对转录组主要是基因表达量的分析。以基因组为参考,除了可以获得基因表达量信息之外,还可以进⾏可 变剪切、突变位点、融合基因等基因结构研究,这对⾮模式⽣物的基因组优化或临床肿瘤等研究具有重要的意义。
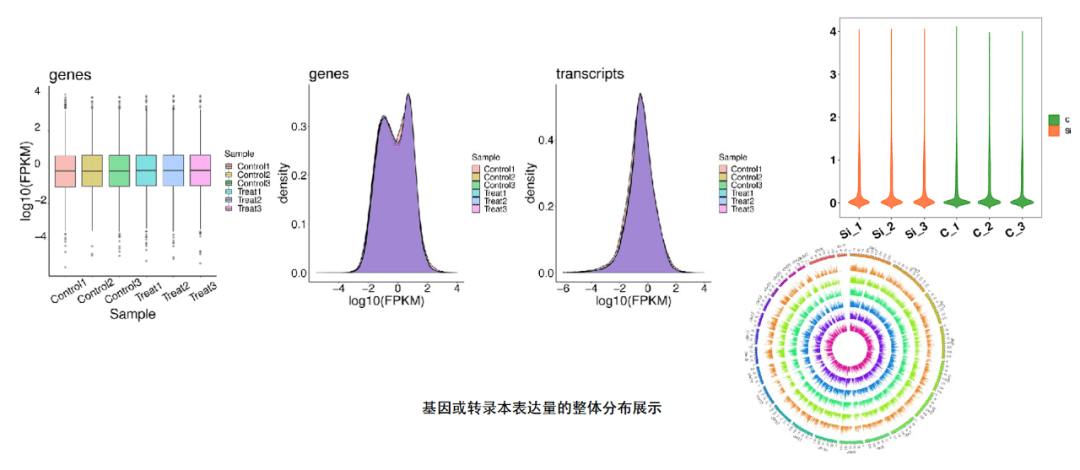
⼀个基因表达⽔平的直接体现就是其转录本的丰度情况,转录本丰度程度越⾼,则基因表达⽔平越⾼。针对转录组我们提供两个维度的定 量,即基因层⾯和转录本层⾯。基因表达量的计算使⽤FPKM(Fragments per kb per Million reads)值,即每百万fragments中来⾃某⼀基因每千碱基⻓度的fragments数⽬。
FPKM同时考虑了测序深度和基因⻓度对fragments计数的影响,是⽬前最为常⽤的基因表达⽔平估算⽅法 之⼀。FPKM法能消除基因⻓度和测序量差异对计算基因表达的影响,计算得到的基因表达量可直接⽤于⽐较不同样品间的基因表达差异。
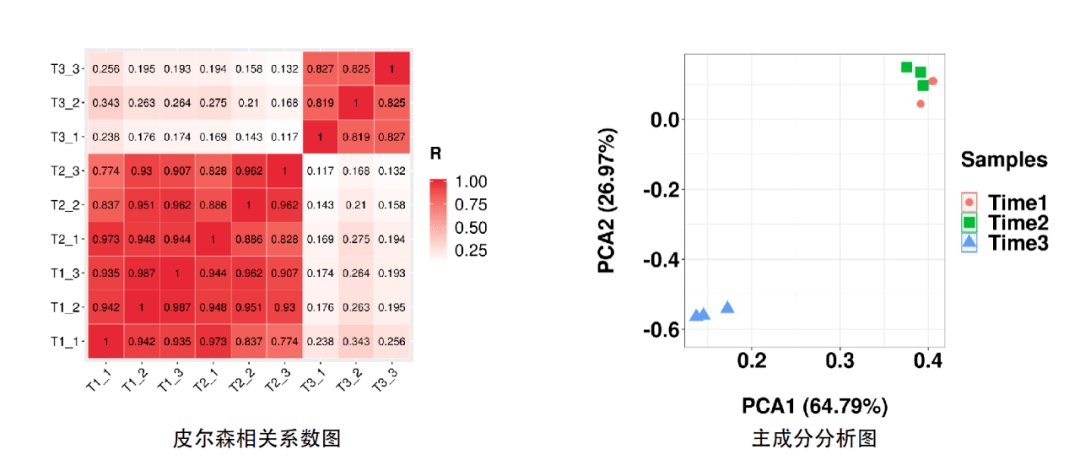
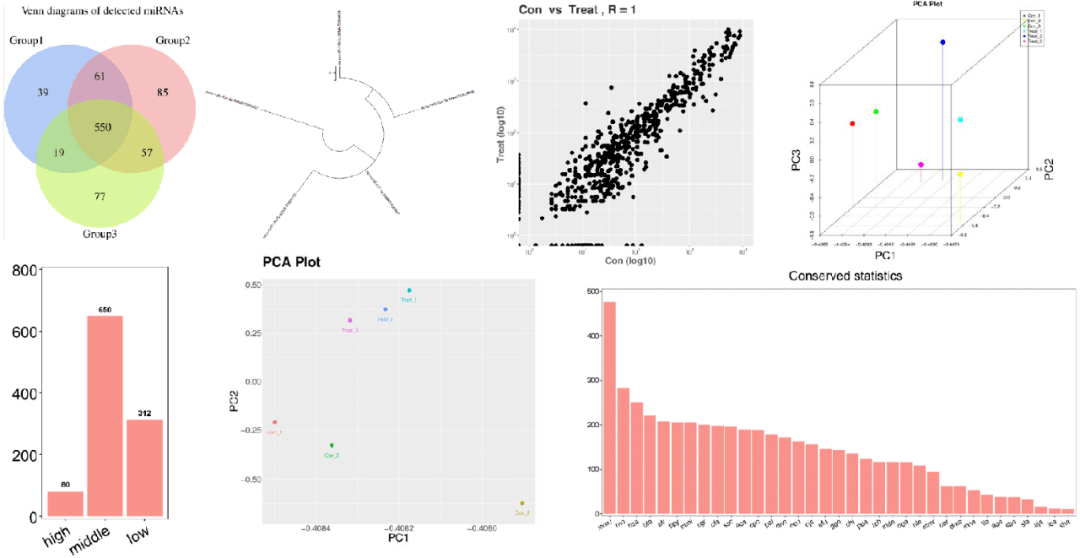
基于样本基因表达量,通过主成分分析(PCA)和计算样本间pearson相关系数,了解样本之间的重复性情况和组间差异情况,辅助于排除离群样本。
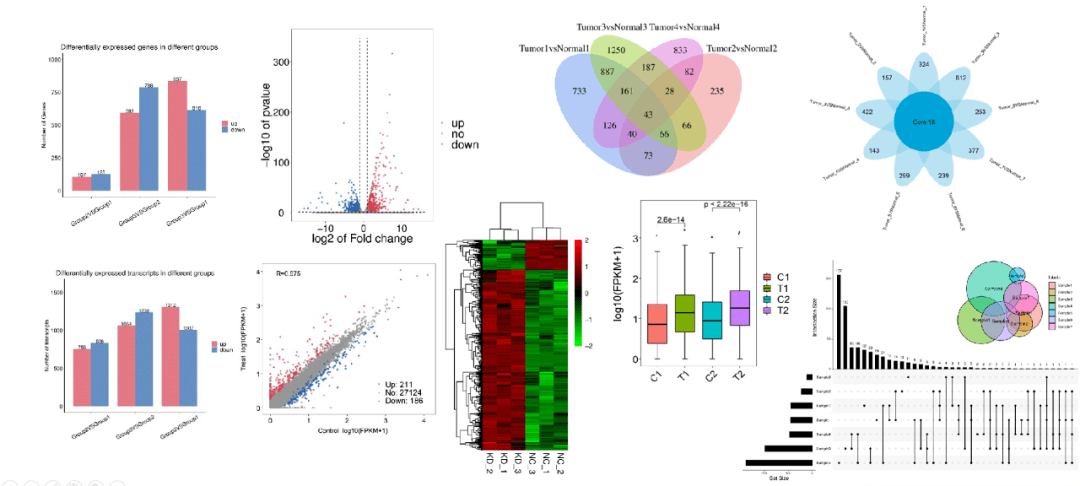
使⽤edgeR或DESeq2分析组间差异显著基因或转录本,差异显著的阈值为差异倍数>2或<0.5且pvalue < 0.05(或FDR < 0.05)。差异结果呈现形式包括火山图、散点图、UpSet图、热图、Venn图、花瓣图等。
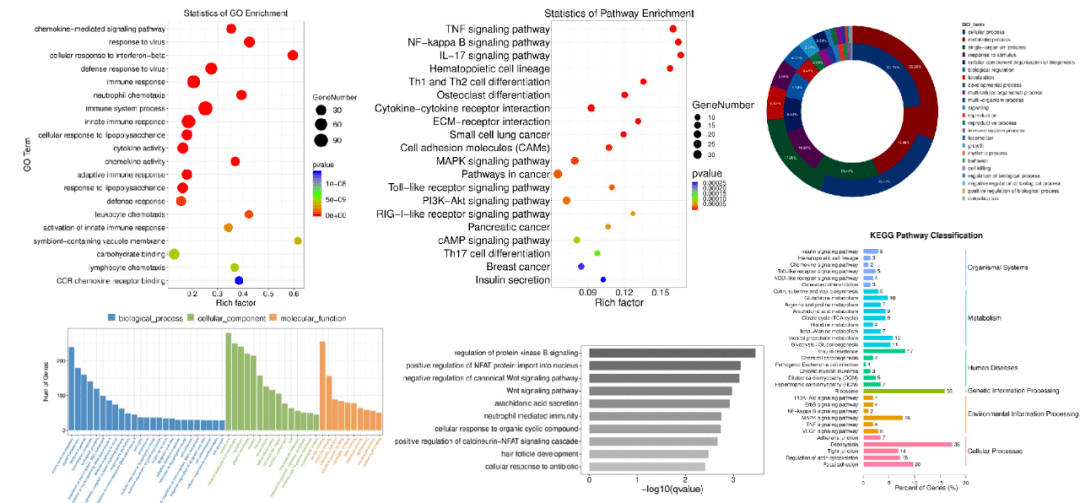
GO和KEGG功能富集分析除了传统的散点图和柱状图外,还加入了广受好评颜值颇高的圈层图和即将加入的雷达图。

除了常规的基因和分子之间的调控网络Cytoscape图外,还特意加入了针对不同信号通路 p value的热图,将您的分析再次升级。
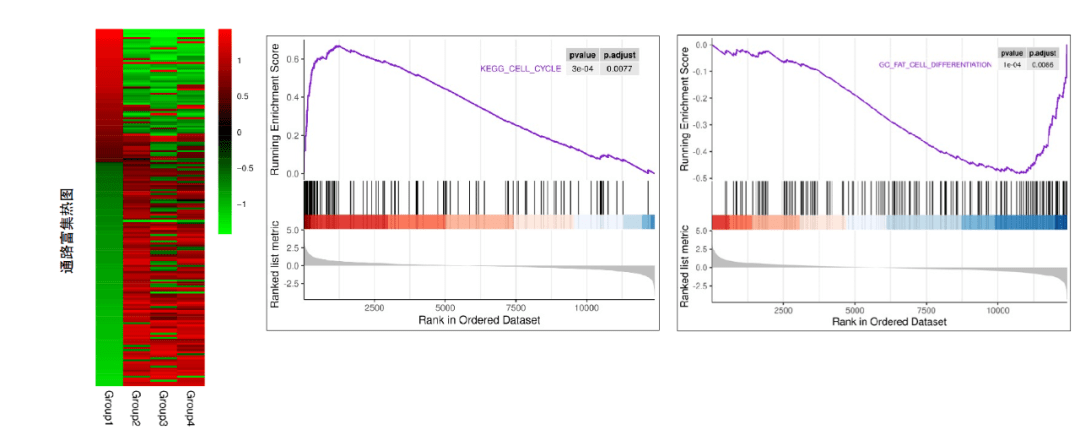
GSEA(Gene Set Enrichment Analysis),也叫基因集富集分析。其基本思想是使⽤预定义的基因集,将基因按照在两类样本中的差异表达程度排序,然后检验预先 设定的基因集合(Gene Set)是否在这个排序表的顶端或者底端富集。
基于传统的超⼏何检验的富集分析往往需要⽤到显著差异基因集数据,当单个基因变化较为微弱时,基于传统富集分析得到结果可能会很少,甚⾄没有结果。
GSEA分析能够有效弥补传统富集分析对微效基因的有效信息挖据不⾜等问题,更为全⾯地对某⼀功能单位(KEGG通路、GO条⽬或其他)的调节作⽤进⾏解释。
不过该分析推荐物种为人和小鼠,其他物种由于注释信息不全,gene sets信息不完整,无法开展进一步的GSEA分析。
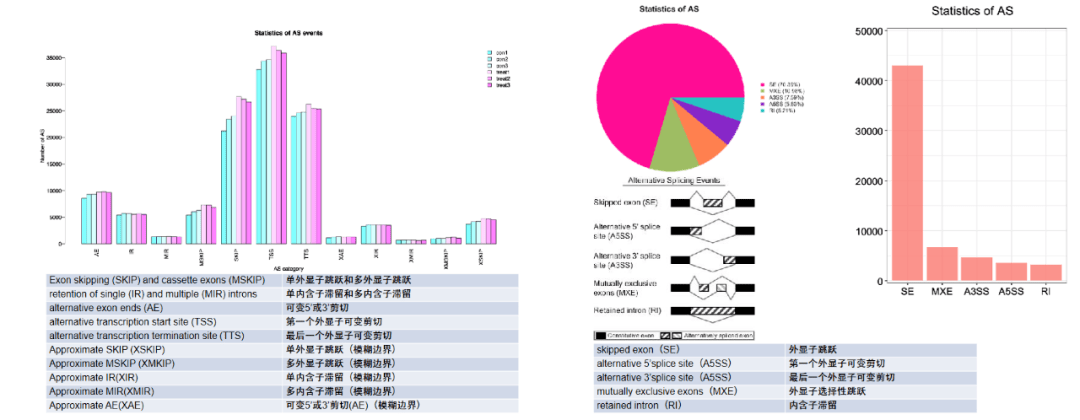
可变剪切(differential splicing),也叫做选择性剪切(alternative splicing),指的是在mRNA前体到成熟mRNA的过程当中,不同的剪切⽅式使得同⼀个基因可以产⽣多个不同的成熟mRNA,最终产⽣不同的蛋⽩质(蛋⽩编码基因座可以通过可变剪切产⽣⾮编码RNA,⽐如lncRNA;RNA基因可以通过可变剪切产⽣不同的⾮编码RNA)。可变剪切在真核⽣物体内⼴泛存在,有研究指出,对于⼈类基因组中包含多个exon的基因⽽⾔,绝⼤部分都存在可变剪切现象。可变剪切导致了转录本和 蛋⽩质结构与功能的多态性,是⼀种重要的转录调控机制。
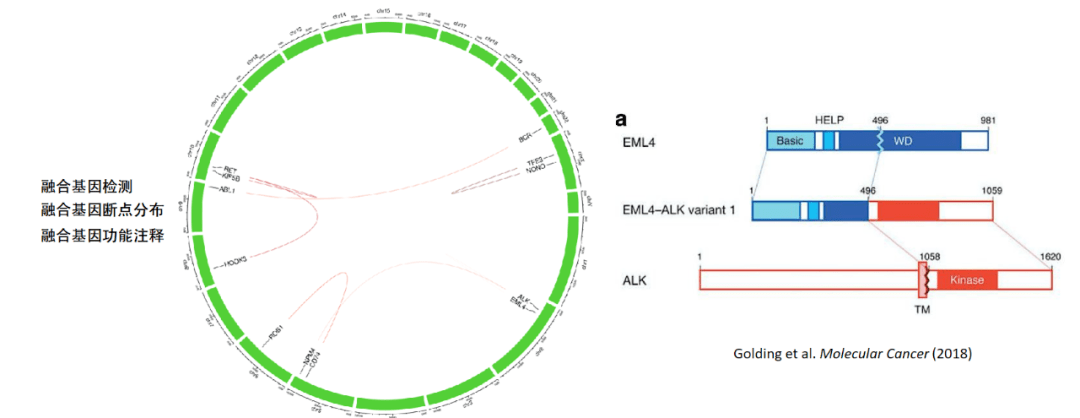
融合基因可以导致异常序列或功能蛋⽩的产⽣,常与肿瘤发⽣发展相关,如常⻅的EML4-ALK、BCR-ABL、CD74-ROS1融合基因等。基于star-fusion可以检测基因融合事件,对于临床肿瘤样本具有重要的参考意义。
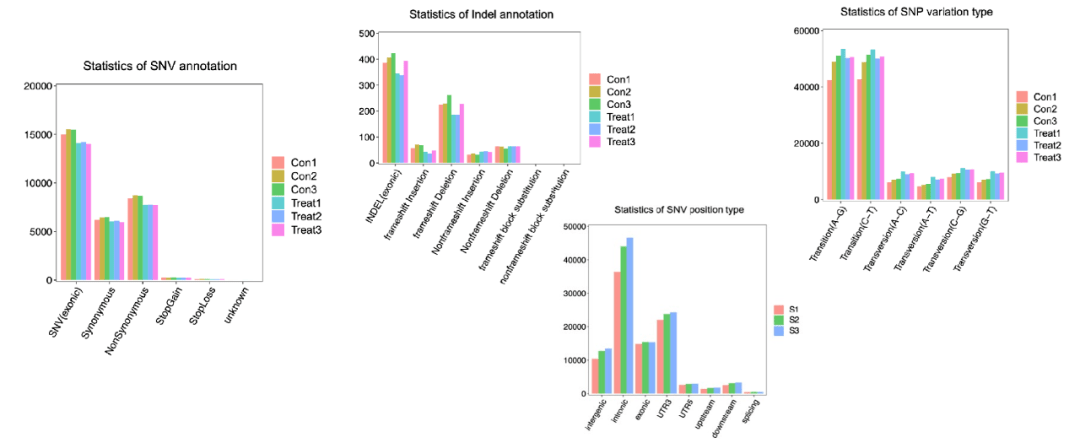
SNP(单核苷酸多态性)是指在基因组上单个核苷酸的变异,其数量很多,多态性丰富。基因组上单个核苷酸的变异包括置换、颠换、缺失 和插⼊。根据单核苷酸碱基形态的多样性,可以分为置换(transition,CT,在其互补链上则为GA)和颠换(transversion,CA,GT,CG,AT)。InDel(insertion-deletion)是指相对于参考基因组,样本中发⽣的⼩⽚段的插⼊缺失,该插⼊缺失可能含⼀个或多个碱基。
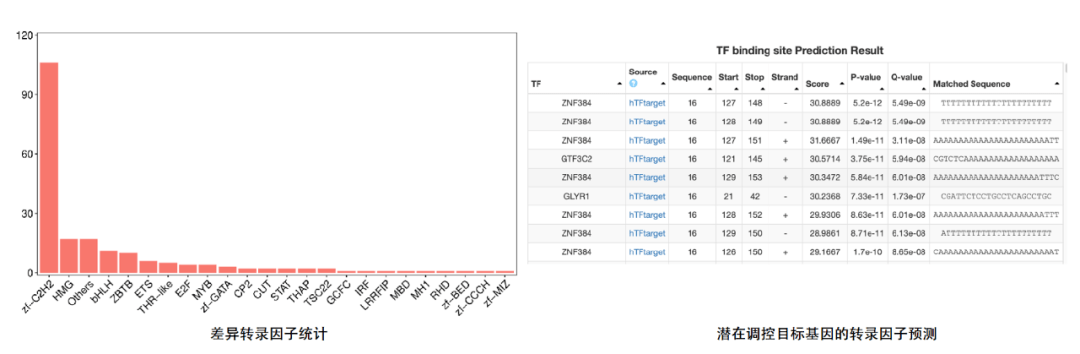
转录因⼦(Tranion Factors, TFs)是指能够以序列特异性⽅式结合DNA并且调节基因转录的蛋⽩质。通过将转录组测序检测到的基因与转录因⼦数据库进⾏映射(动物是AnimalTFDB,植物是PlantTFDB),可以获得转录因⼦注释,结合基因定量和差异分析可以获取转录因⼦ 的表达量信息和差异信息。结合在线⽹站和基因共表达分析,可以预测可能调控感兴趣基因的转录因⼦。
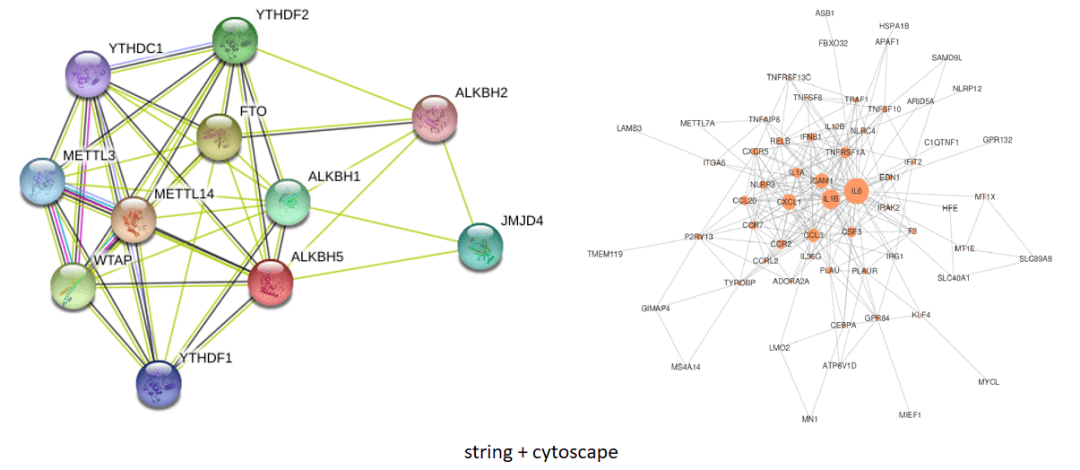
利⽤string蛋⽩质互作数据库(hep://string-db.org)中的互作关系进⾏差异基因蛋⽩互作⽹络(PPI)分析,利⽤cytoscape(heps://cytoscape.org)构建互作关系⽹络图。针对数据库中不包含的物种,则利⽤blastx将⽬标基因序列⽐对到string数据库中包含的参考物种的蛋⽩质序列上,并利⽤该参考物种的蛋⽩质互作关系构建互作⽹络。
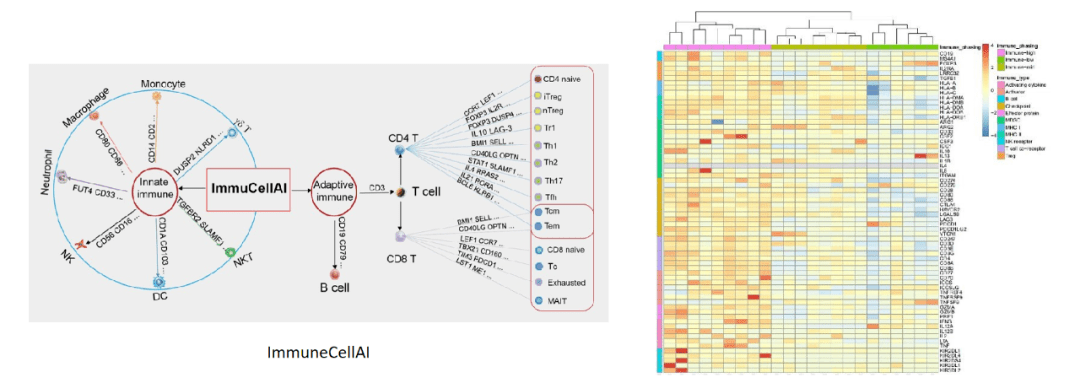
基于免疫相关基因进⾏聚类分析,可以进⾏免疫分型分析。利⽤去卷积等⽅法可以基于转录组表达谱数据估算免疫细胞(B细胞、NK细胞、巨噬细胞等)的丰度。
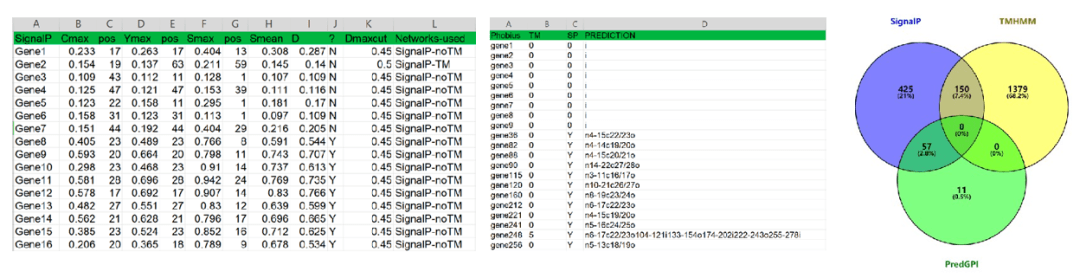
基于检测到的基因序列可以预测其ORF或翻译潜能,获得氨基酸序列(或直接从参考基因组获取氨基酸序列),然后通过预测信号肽、跨膜 螺旋等预测蛋⽩的亚细胞定位和预测分泌蛋⽩。
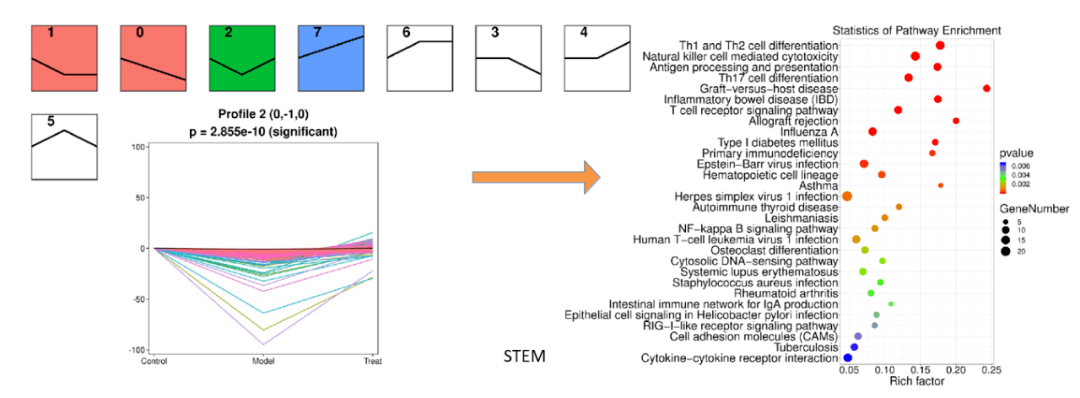
表达模式⼀般指基因表达量随时间点变化的规律。⽐如研究药物对疾病模型的治疗作⽤时,⼀般分为经典的对照组、模型组(造模)和模 型⽤药组(或患病、治疗和治愈),我们希望看到药物治疗对于模型是有效的,那么与此相关的表达模式包括先上调后下调和先下调后上 调。⼀般⽽⾔符合某种特定模式的基因可能是参与相同的代谢通路,也可能是受到了相同分⼦的调控。对于特定表达模式下的基因进⾏GO、KEGG功能富集分析,有助于挖掘潜在规律。与传统差异分析不同的是,基因表达模式聚类分析中更关键的是筛选感兴趣的表达模式,然后对其模式下的基因进⾏后续的功能分析。
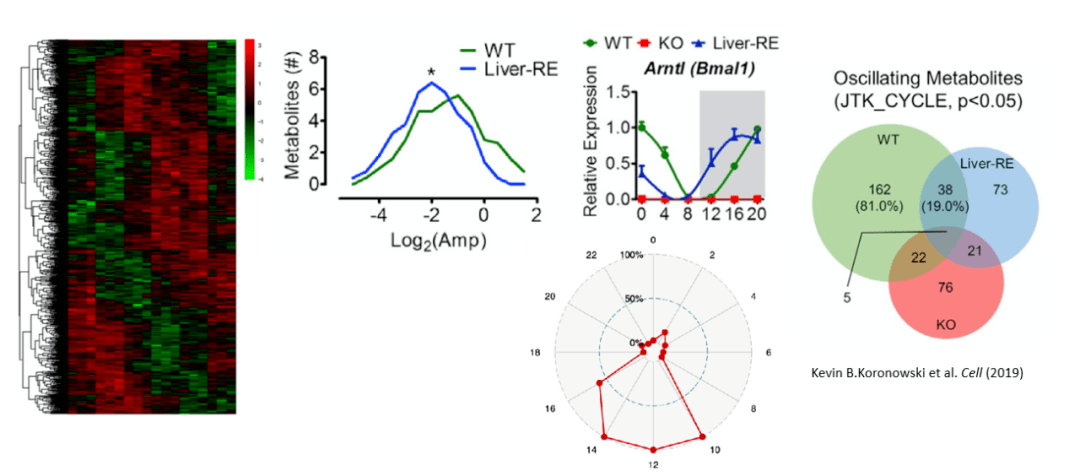
研究表明,在脊椎动物中,⽣物钟分⼦调控⽹络⼏乎存在于每种组织和细胞类型中。外周⽣物钟的发现,让⼈们意识到外周⽣物钟似乎具 有⼀定的⾃主节律性,各个外周⽣物钟之间的相互作⽤将在⽣理机能和疾病发⽣过程中起着关键作⽤。JTK_CYCLE是⼀种⾮参数测试程序, 可从微阵列数据或转录组数据中检测节律转录本。
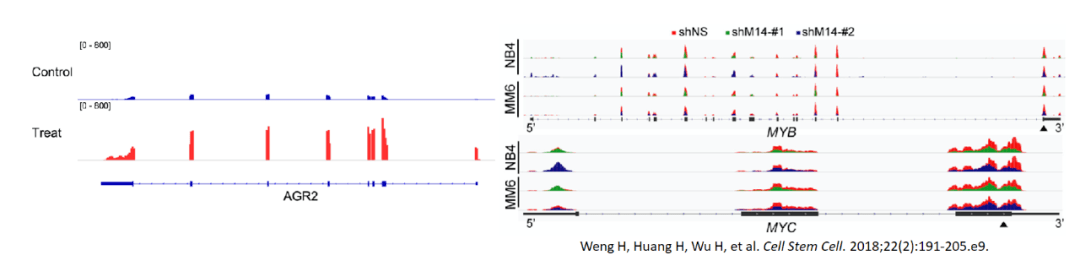
使⽤整合基因组浏览器(Integrative Genomics Viewer,IGV)可以进⾏基因组数据的可视化,直观展示基因组及其各种注释信息,也可以直观从覆盖度上⽐较基因的表达量、m6A或DNA甲基化修饰区域等。IGV官⽹为hep://www.igv.org。
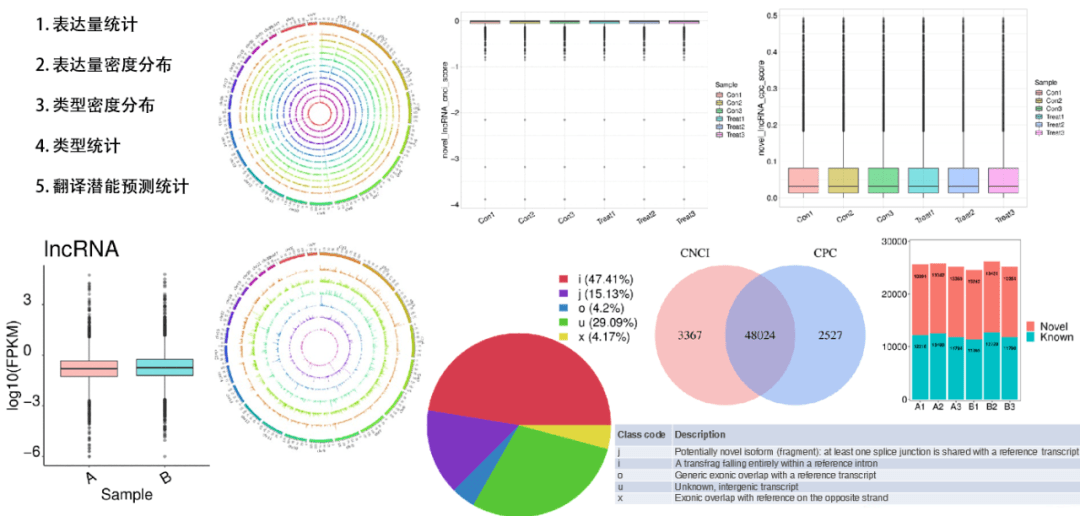
lncRNA统计学类分析五件套:表达量统计、表达量及lncRNA类型circos图、lncRNA不同类别分类统计以及lncRNA翻译潜能分析。
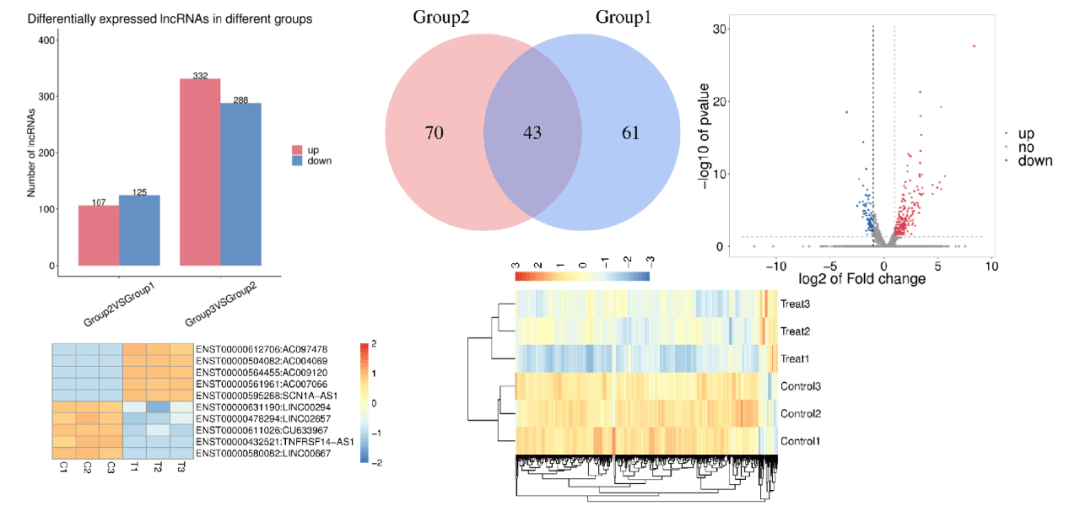
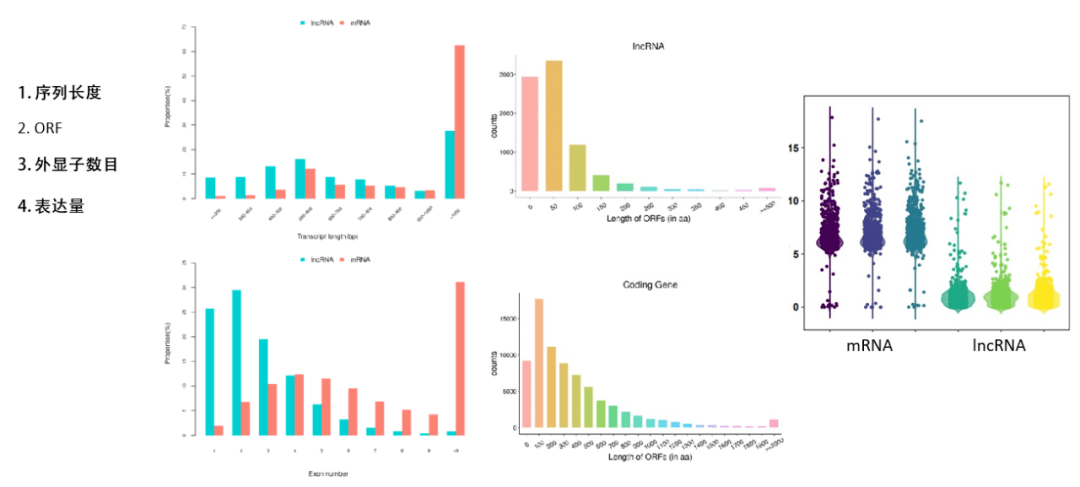
使⽤edgeR或DESeq2分析组间差异显著基lncRNA,差异显著阈值为差异倍数>2或<0.5且pvalue < 0.05(或FDR < 0.05)。
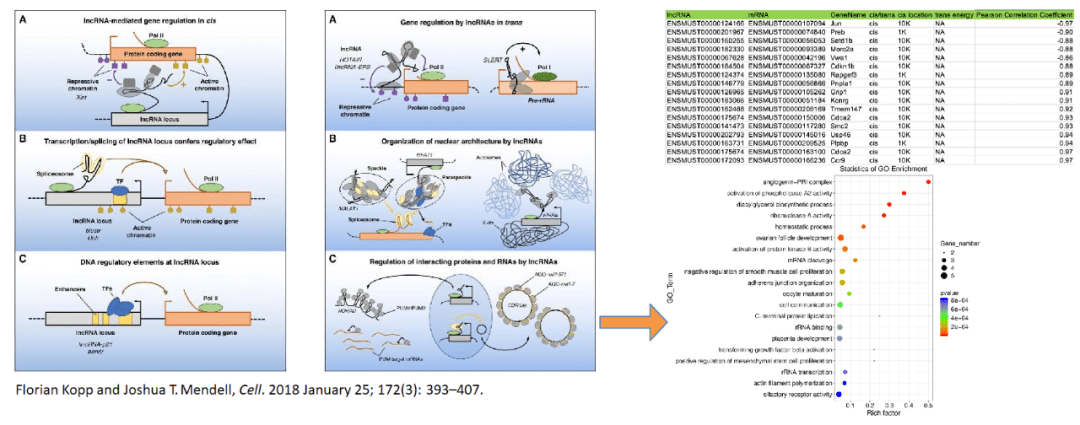
顺式调控(cis regula^on)即lncRNA调控与其邻近基因的表达。lncRNA顺式调控靶基因主要根据位置关系预测,定义染⾊体中上下游100kb 范围内存在的差异表达的lncRNA与差异表达的mRNA可能构成顺式调控,并基于表达量计算mRNA和lncRNA的相关性(Pearson Correla^on Coefficient)。反式调控(trans regula^on)即lncRNA跨染⾊体调控基因的表达,trans调控靶基因主要基于lncRNA与mRNA序列之间形成⼆级 结构所需要⾃由能的⼤⼩,如果两条序列需要很低的⾃由能就可以结合,则两者间可能存在相互作⽤。通过顺式/反式可以预测lncRNA的靶 基因,然后可以对靶基因进⾏功能富集分析。

lncRNA的亚细胞定位和其功能与作⽤机制是相关的,可以使⽤在线⽹站查询或预测lncRNA的亚细胞定位,获得潜在功能机制的参考。软件预测仅供参考,需要FISH等实验验证其定位。
RNALocate: http://www.rna-society.org/rnalocate/
lncATLAS: http://lncatlas.crg.eu/
lncLocator: http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/
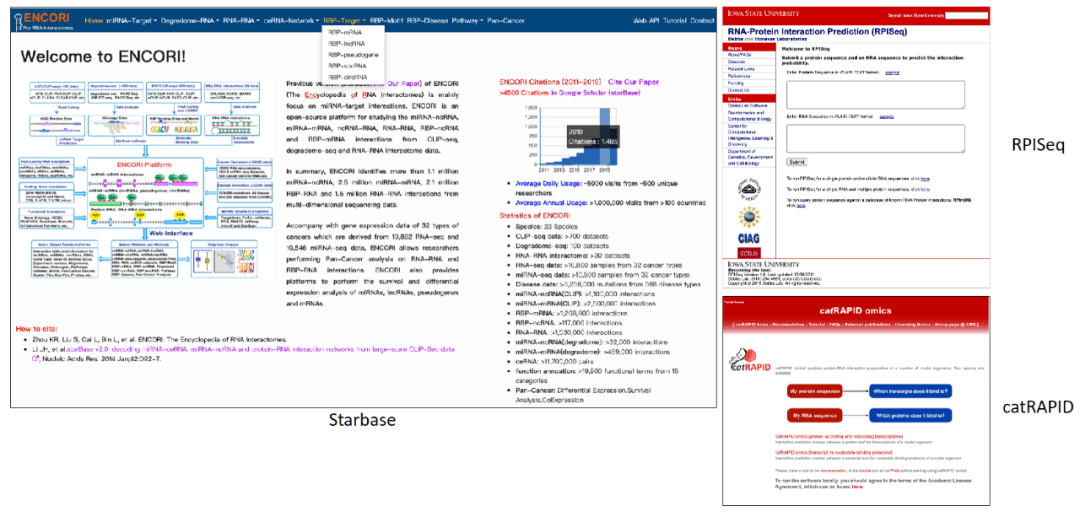
lncRNA可以作为功能复合体⽀架⽽发挥作⽤,通过在线⽹站查询和预测lncRNA互作蛋⽩可以为lncRNA功能机制研究获得更多参考信息。此 外可通过RNA pull down(结合免疫印迹或质谱)、RIP-seq等⽅法检测感兴趣RNA结合蛋⽩(RBPs)结合的lncRNA或关注lncRNA结合的蛋⽩。
Starbase: http://starbase.sysu.edu.cn/
RPISeq: http://pridb.gdcb.iastate.edu/RPISeq/
catRAPID: http://service.tartaglialab.com/page/catrapid_omics_group
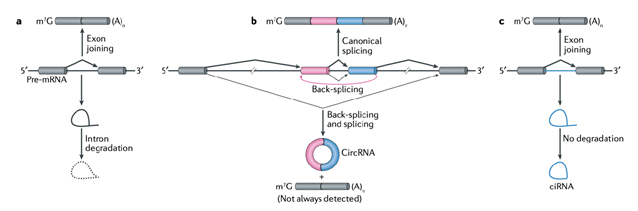
环状RNA(circRNA)是⼀类新的具有调控功能的⾮编码RNA,具有闭合环状结构⼤量存在于真核转录组中。⼤部分的环状RNA是由外显⼦序列构成,在不同的物种中具有保守性,同时存在组织及不同发育阶段的表达特 异性。由于环状RNA对核酸酶不敏感,所以⽐线性RNA更为稳定,这使得环状RNA在作为新型临床诊断标记物的 开发应⽤上具有明显优势。
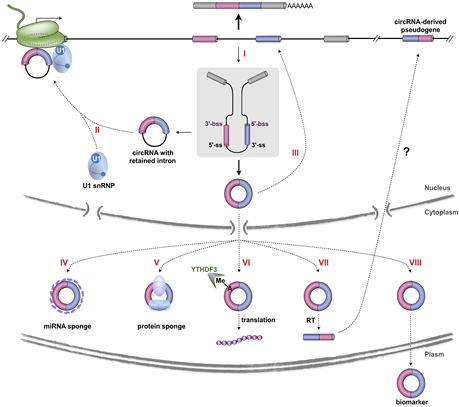
内源性circRNA通过不同的机制参与基因表达调控:
1:circRNA的加⼯会影响其线性mRNA的剪接;
2:circRNA可以调节其亲本基因的转录;
3:circRNA可以调节其线性同源物的剪接;
4:circRNA可以充当miRNA海绵;
5:circRNA可以通过相关蛋⽩起作⽤;
6:circRNA可以翻译;
7:circRNA是衍⽣假基因的资源;
8:circRNA是有前景的⽣物标志物。
circrRNA涉及大量高分研究,一直是国自然和高分杂志的热点非编码RNA分子之一。
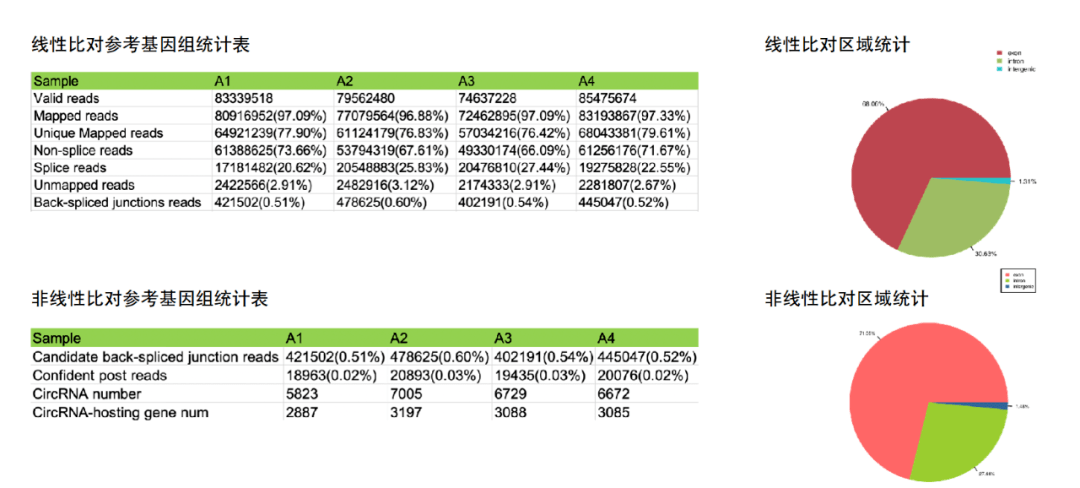
使⽤hisat2、tophat和tophat fusion⽐对参考基因组,基于线性⽐对进⾏线性转录本的定量,基于⾮线性⽐对进⾏circRNA的鉴定和定量。
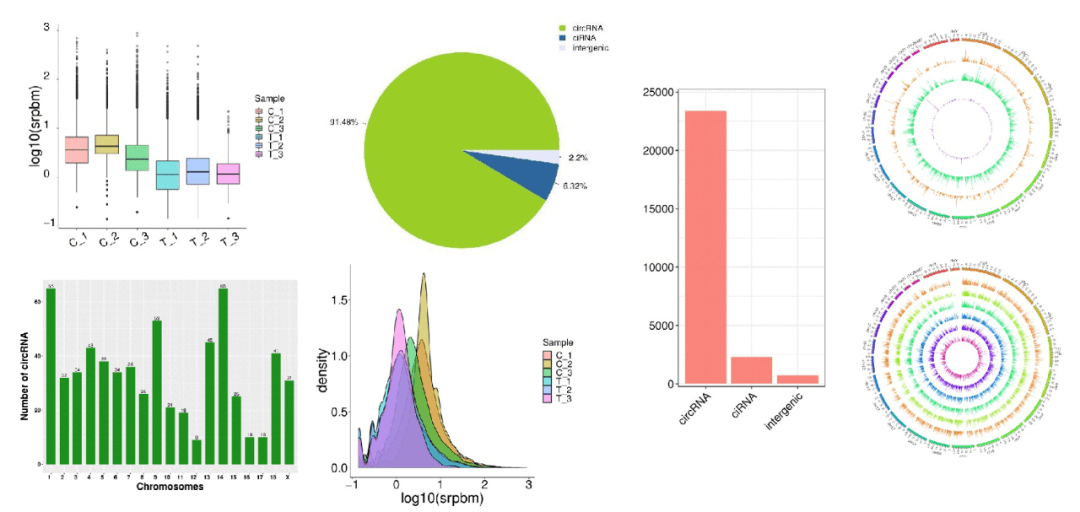
circRNA鉴定最关键的原理是寻找反向剪接的reads,即backsplice reads。使⽤CIRCExlorer2和CIRI进⾏circRNA的鉴定,使⽤srpbm(spliced reads per billion mapping)进⾏circRNA的定量。srpbm = number of circular reads / (number of mapped reads (units in billion) * read length)
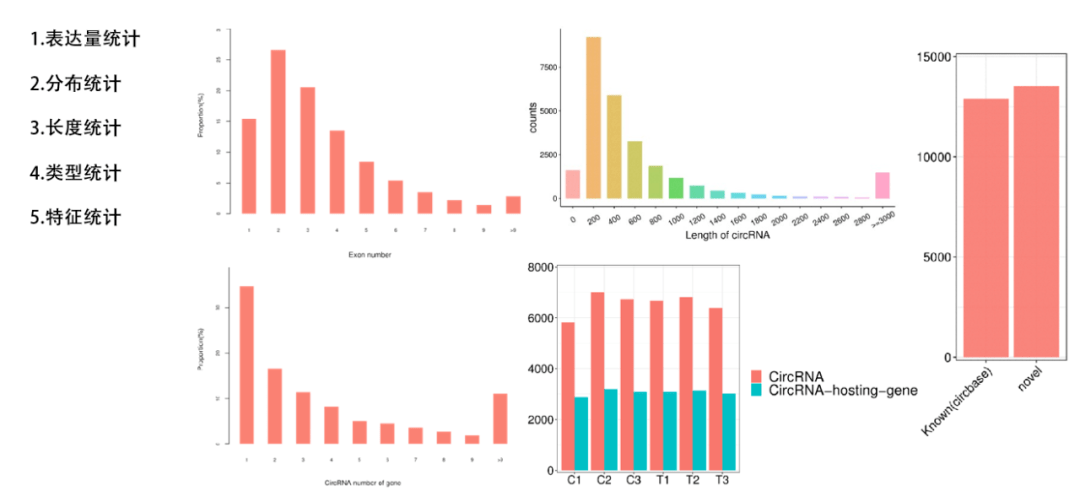
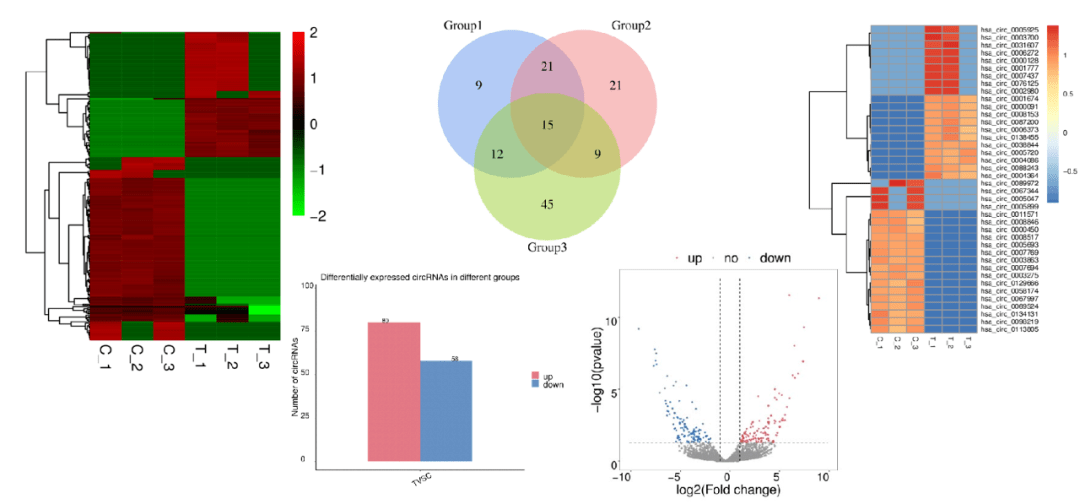
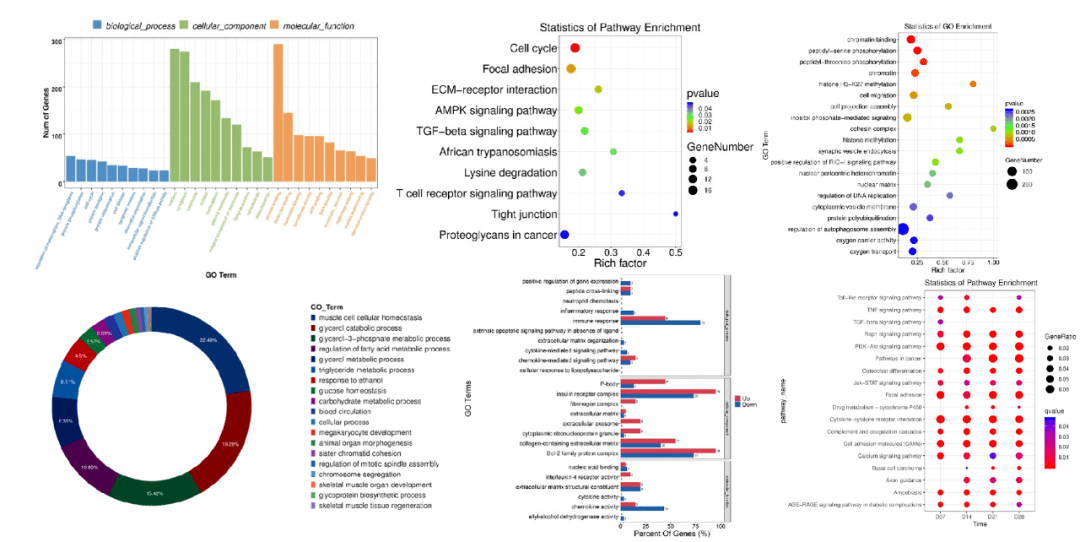
使⽤edgeR分析组间差异circRNA,差异显著的阈值为差异倍数>2或<0.5且pvalue < 0.05。并对circRNA所在的gene的GO和KEGG注释进行富集分析。

通过在线⽹站查询和预测circRNA互作蛋⽩可以为circRNA的功能机制研究获得更多参考信息。此外可以通过RNA pull down(结合免疫印迹或质谱)、RIP-seq等⽅法检测感兴趣RNA结合蛋⽩(RBPs)结合的circRNA或关注circRNA结合的蛋⽩。
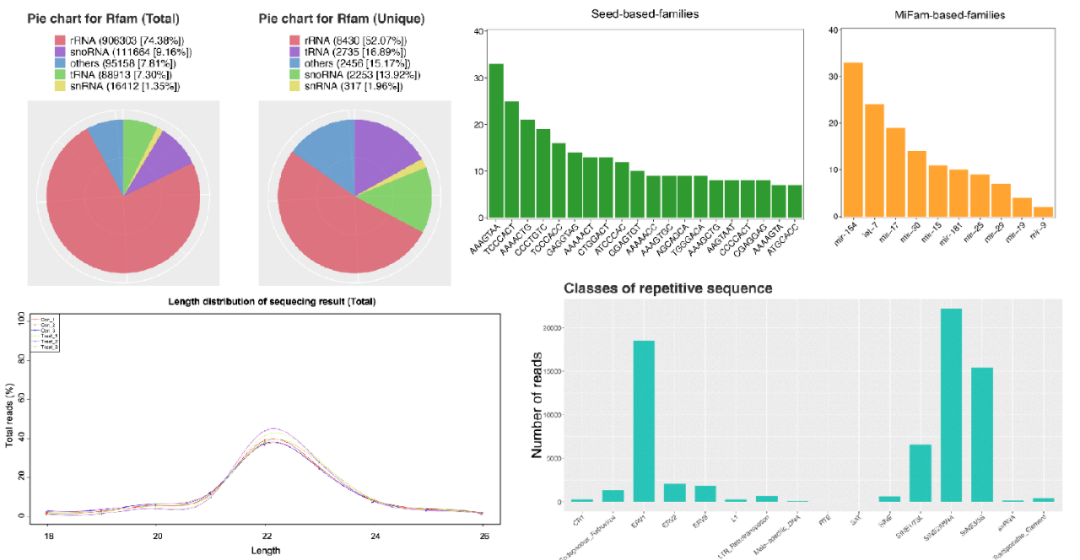

根据实验设计不同采⽤适合的差异检验⽅法筛选差异显著miRNA,例如Fisher精确检验、卡⽅(2*2)、卡⽅(N*N)、T检验以及 ANOVA等算法。对于有⽣物学重复的样本分析,对差异表达miRNA的筛选以pvalue < 0.05为阈值;⽽对⽆⽣物学重复的样本分析,对差异表达miRNA的筛选以|log2(Foldchange)| ≥ 1(即两倍差异倍数)且pvalue < 0.05为阈值。
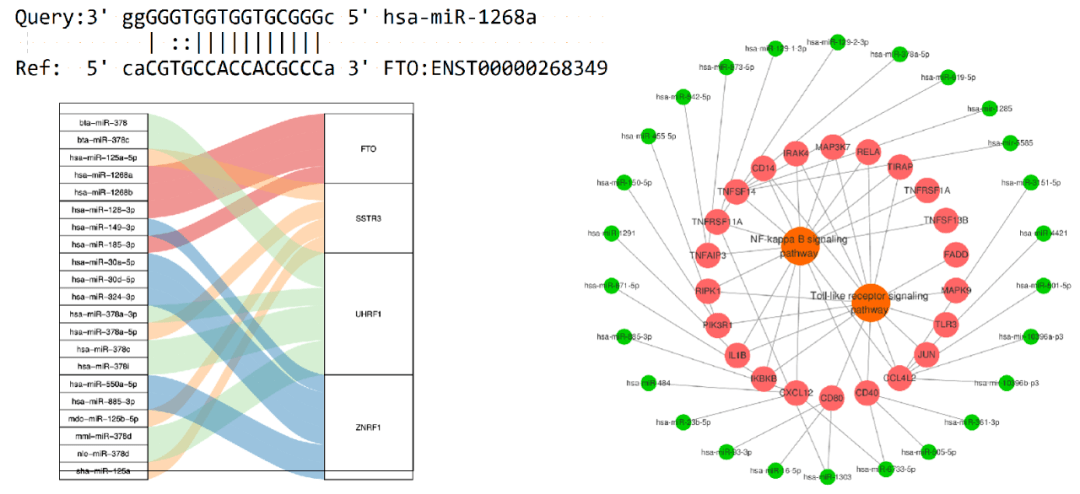
针对动物物种,使⽤TargetScan、miRanda两款软件对显著性差异miRNA分别进⾏靶基因预测,对两款软件预测出的靶基因分别按照每款软件的评分标准进⾏筛选并取两款软件预测的交集作为差异miRNA的最终靶基因。针对植物物种使⽤CleaveLand4:GSTAr对显著性差异miRNA进⾏靶基因预测。
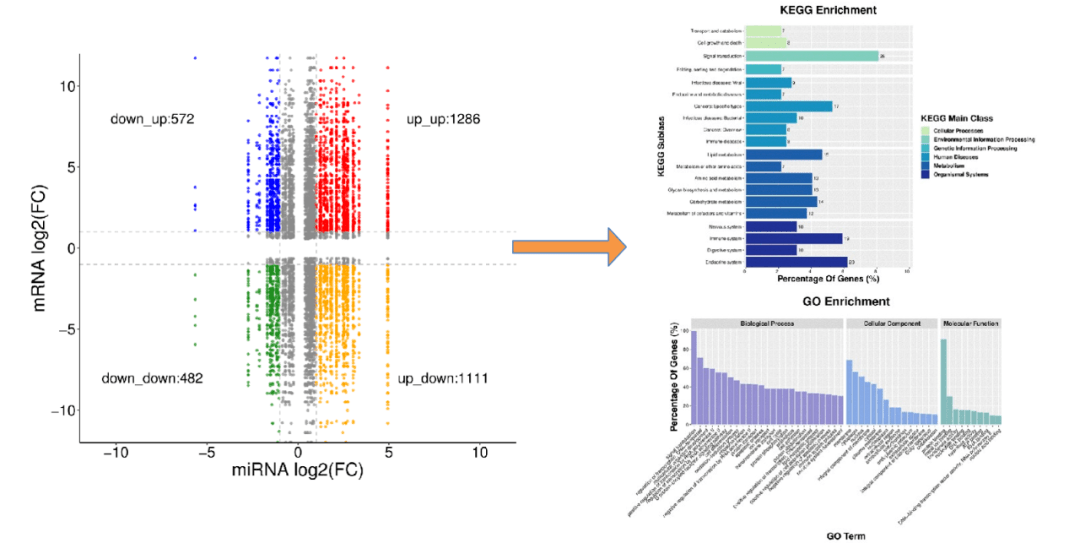
基于miRNA靶基因预测和miRNA和mRNA的变化趋势可以将存在靶向的miRNA-mRNA关系对进⾏分类,主要分为up_up、down_down的正相关 和up_down、down_up的负相关,然后对每类中的mRNA进⾏功能富集分析。
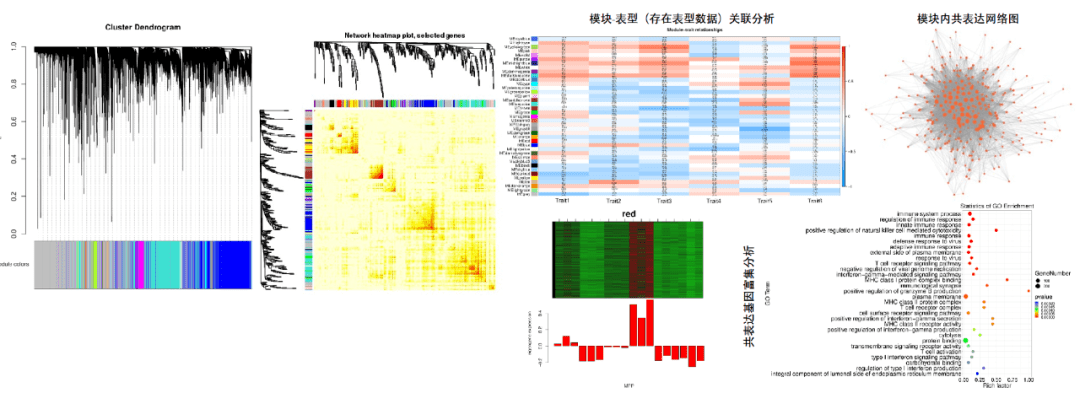
在⼤样本中,差异分析或趋势分析⽆法对基因进⾏有效分类,且基于数据的功能分析⽆法推断新的调控关系。利⽤WGCNA(加权基因共表达⽹络分析)可以将表 达模式相似的基因(蛋⽩编码基因或RNA基因)进⾏聚类,构建共表达⽹络。WGCNA旨在寻找协同表达的基因模块(module),并探索基因⽹络与关注表型之间 的关联关系,以及⽹络中的核⼼基因(Hub基因)。WGCNA适⽤于复杂的数据模式,⼀般可应⽤的研究⽅向有:不同器官或组织类型发育调控、⾮⽣物胁迫不同 时间点应答、病原菌侵染后不同时间点应答等。WGCNA中两个关键的概念是模块与核⼼基因。模块是⾼度内连的基因集。在⽆向⽹络中,模块内是⾼度相关的基 因。在有向⽹络中,模块内是⾼度正相关的基因。样本数n ≥ 15 时可以进⾏WGCNA分析;样本数 6 ≤ n < 15 时,则利⽤⽪尔森相关系数构建基因的共表达关系。
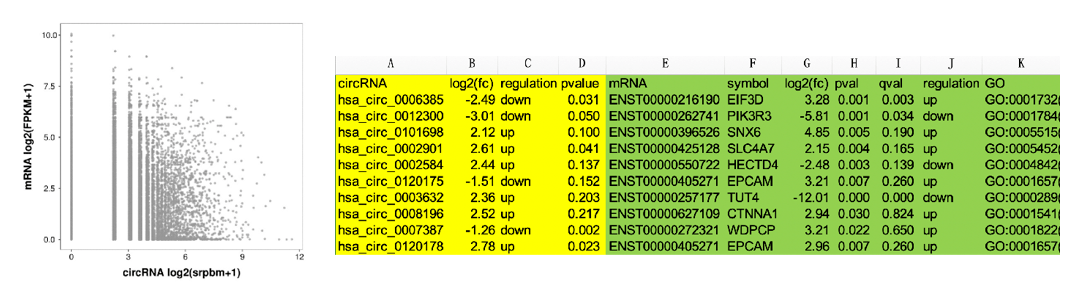
基于circRNA和亲本基因(mRNA)的对应关系可以进⾏两组学的关联分析,探究circRNA与亲本基因(mRNA)的表达量相关性和变化趋势相关性。
不同于动物,植物miRNA通常与靶基因进⾏完全或接近完全的配对引起靶基因的剪切从⽽调控基因的表达,基于这⼀特征可以进⾏植物miRNA靶基因的⽣物信息学预测。然⽽,由于预 测⽅法⽆法区分预测靶基因的真伪,所有预测结果必须经实验证实以去伪存真,极⼤地影响了靶基因的探索效率。随着⾼通量测序技术的出现和发展,结合⾼通量测序技术和⽣物 信息学各⾃优势的降解组测序(Degradome Sequencing)可以使科研⼈员摆脱了⽣物信息学预测的限制,真正从实验中找到了miRNA的作⽤靶基因。
- 本文固定链接: https://maimengkong.com/zu/1125.html
- 转载请注明: : 萌小白 2022年7月16日 于 卖萌控的博客 发表
- 百度已收录
