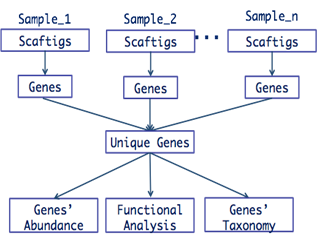
在前两期的推送稿中,我们为大家介绍了宏基因组组装的基本原理和操作。基于组装序列,我们可以实现基因预测、物种注释、功能注释等相关分析,从而研究微生物菌群结构、菌属功能及作用机制。因此,本期我们将从基因预测的原理和操作两个部分出发,为大家介绍基于组装序列的基因预测。
1
基因预测原理
宏基因组基因预测一般包括同源预测和从头预测。同源预测是通过与基因的同源序列比对,从而获得与已知基因序列最大匹配,其预测依赖于已知的基因信息,且不能注释出在数据库中缺少功能相似性序列的基因和新基因,计算资源消耗过大,时间花费过长。而从头预测是根据给定的序列特征来预测,主要依赖于在编码区和非编码区拥有不同特征的信息,并在统计学上进行描述,构建概率模型,用以区别编码与非编码区。从头预测能够预测出已知的和未知的基因,且计算资源消耗小,时间花费少,常用软件包括:GeneMark,MetaGeneMark,MetaGene等。本期我们主要通过基于从头预测原理的MetaGeneMark来预测基因。
预测过程包括

2
基因预测实现
(1)软件
MetaGeneMark(预测的范围是细菌和古菌),下载地址:
http://exon.gatech.edu/license_download.cgi。
CD-HIT去除冗余序列,下载地址:http://www.bioinformatics.org/cd-hit。
(2)输入文件
SOAPdenovo-63mer对单个样本进行组装后,筛选出长度不小于500bp的scaftigs,得到结果文件sample1.cut500.scafSeq,sample2.cut500.scafSeq,sample3.cut500.scafSeq;SOAPdenovo-63mer对所有样本进行混合组装,筛选出长度不小于500bp的scaftigs,得到结果文件mix.cut500.scafSeq。文件格式如图所示,包括scaffold编号,长度及序列信息。

(3)基因预测
基于单个样本的基因预测
gmhmmp -a -d -f G -m MetaGeneMark_v1.modsample1.cut500.scafSeq -A sample1_protein.fasta -D sample1_nucleotide.fasta
gmhmmp -a -d -f G -m MetaGeneMark_v1.modsample2.cut500.scafSeq -A sample2_protein.fasta -D sample2_nucleotide.fasta
gmhmmp -a -d -f G -m MetaGeneMark_v1.modsample3.cut500.scafSeq -A sample3_protein.fasta -D sample3_nucleotide.fasta
基于混合组装的基因预测
gmhmmp -a -d -f G -m MetaGeneMark_v1.modmix.cut500.scafSeq -A mix_protein.fasta -D mix_nucleotide.fasta
参数说明:
-a 显示预测得到的基因的蛋白序列
-A 蛋白序列输出文件
-d 显示预测得到的基因的核酸序列
-D 核酸序列输出文件
-f显示输出格式,L=LST,G=GFF
-m 用于基因预测的模型文件,MetaGeneMark提供的MetaGeneMark_v1.mod适用于宏基因组预测
(4)基因去冗余
A. 将上一步得到的所有核酸序列(sample1_nucleotide.fasta,sample2_nucleotide.fasta,sample3_nucleotide.fasta,mix_nucleotide.fasta)合并成一个核酸序列total.gene.nucl.fasta; 将所有蛋白序列合并成一个total.gene.prot.fasta。
B. 蛋白质序列去冗余:
cd-hit-c 0.9 -n 5 -M 1600 -d 0 -T 8 -i total.gene.prot.fasta -o NonRundant.gene.prot.fasta
参数说明:
-c 相似性阈值,默认值为0.9
-n word长度值,基于word filter方法使用不同word长度值产生的去冗余水平不同,例如长度为2的word得到相似性在50%以上的序列,长度为3的word得到相似性在66.7%以上的序列
-M 分配的内存
-d 聚类信息文件中序列名长度,0代表显示完整序列名。
-T 线程数
-i 输入文件,fasta格式
-o 输出文件前缀,有两个输出文件,分别为fasta格式和以.clstr结尾的聚类信息文件。
输出文件:去冗余后的fasta格式文件NonRundant.gene.prot.fasta;以.clstr结尾的聚类信息文件NonRundant.gene.prot.fasta.clstr(见下图)。
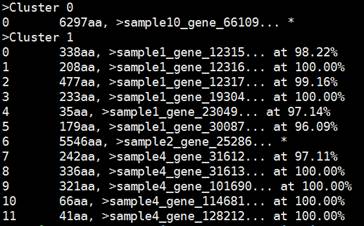
注:每一个聚类组以“>”区分,且每个聚类组有不同的聚类序列,每个聚类序列中百分比代表该序列与代表序列的相似度,“*”代表该序列即为代表序列。
C. 核酸序列去冗余:
由于不同的核酸序列翻译后可能产生相同的蛋白质序列,因此对核酸序列的去冗余需要基于蛋白质序列。具体做法是:基于蛋白质序列文件与核酸序列文件中序列号的对应关系,根据去冗余后的蛋白质序列文件的序列号筛选核酸序列。
(5)基因丰度计算
使用soap将clean data比对到非冗余的核酸序列NonRundant.gene.nucl.fasta。根据soap比对结果,获得比对到某基因的read数。Soap比对工具的使用在之前的微信推送稿中介绍过,这里就不做说明。然后,基于read数获得基因的丰度值,计算方法如下:
|
|
S1 |
S2 |
S3 |
|
G1 |
. |
. |
. |
|
G2 |
. |
. |
. |
|
G3 |
. |
. |
. |
样本i中基因j表示为SiGj, 样本i中比对到基因j的read数表示为SiGj_read,基因j长度表示为Gj_len。
SiGj丰度值=(SiGj_read/Gj_len)/(SiG1_read/G1_len+SiG2_read/G2_len+SiG3_read/G3_len ...)

宏基因组基于组装的基因预测就介绍到这里,大家可以动起手来试一试,在下面一期小编将为大家带来宏基因组物种注释和功能注释部分,敬请期待!
- 本文固定链接: https://maimengkong.com/kyjc/745.html
- 转载请注明: : 萌小白 2021年8月14日 于 卖萌控的博客 发表
- 百度已收录
