解读生信之美,探索每篇文章背后的故事
大家好呀,我是风间琉璃。前面我们学习了这么多转录因子相关的顶级文献操作(“17+干湿结合转录因子生信套路!新的技能点又增加了!国庆回来给老板汇报用!”、“真香预警!近30分多组学干湿结合套路,你看这些图是不是很熟悉,仿佛自己也能来一套的样子?!”、“中科院一区近30分SCI,给你演示下干湿结合顶级套路!学会这个模板,收获一波10+代表作!”),想必大家已经对ChIP-seq和ATAC-seq技术已经不陌生了。
ChIP-seq可能同学们在做实验的时候已经接触到其对应的实验技术ChIP了,那么ATAC-seq具体原理是什么,分析流程是什么呢?今天我们就来学习既往研究者针对ATAC-seq分析流程所发表的综述“From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis”,于2020年发表在《Genome Biology》杂志上。好,废话不多说,我们直接开始解读吧~
虽然前面提到了也要列在这里的传送门
中科院一区近30分SCI,给你演示下干湿结合顶级套路!学会这个模板,收获一波10+代表作!
真香预警!近30分多组学干湿结合套路,你看这些图是不是很熟悉,仿佛自己也能来一套的样子?!
17+干湿结合转录因子生信套路!新的技能点又增加了!国庆回来给老板汇报用!
〇、期刊信息

一、研究背景
ATAC-seq(Assay for Transposase-Accessible Chromatin with high throughput sequencing)于2013年首先开始应用以来,呈指数性增长趋势(Figure 1a)。应用程度比其他DNA开放区域检测技术相比更加普及。由于Tn5转座酶的高反应性,只需要500-5000个细胞就可以进行分析。并且灵敏度和特异度和DNase-seq相当,由于FAIRE-seq,但后两者技术对细胞数的要求更加苛刻。由于ATAC-seq并不需要非常严格的建库标准,所ATAC-seq也能够通过片段的长度识别核小体的位置。并且随着FACS、微流体技术的发展,对于单细胞的scATAC0seq也得到开展,并对揭示临床样本以及发展生物学的细胞异质性具有重要作用。比如正在检测正常造血以及白血病之间染色质可及性的变化。
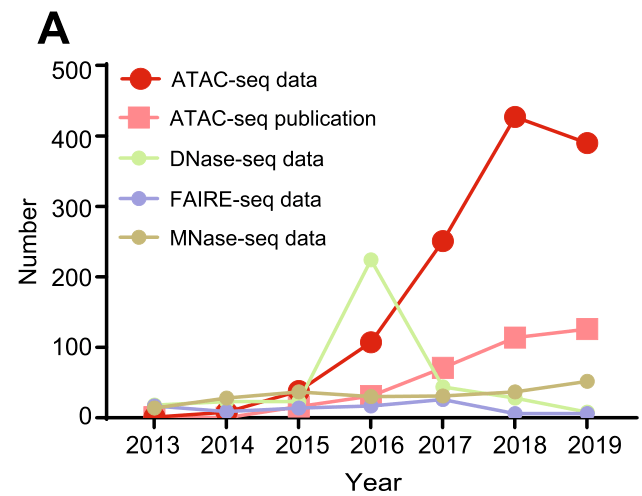
(Figure 1a)
作者根据ATAC-seq数据分为四个步骤:
1.预处理(质控和比对);
2.核心分析(peak鉴定);
3.高级分析(peak注释、motif分析、核小体分析、TF足迹);
4.多组学整合分析(Figure 2)。
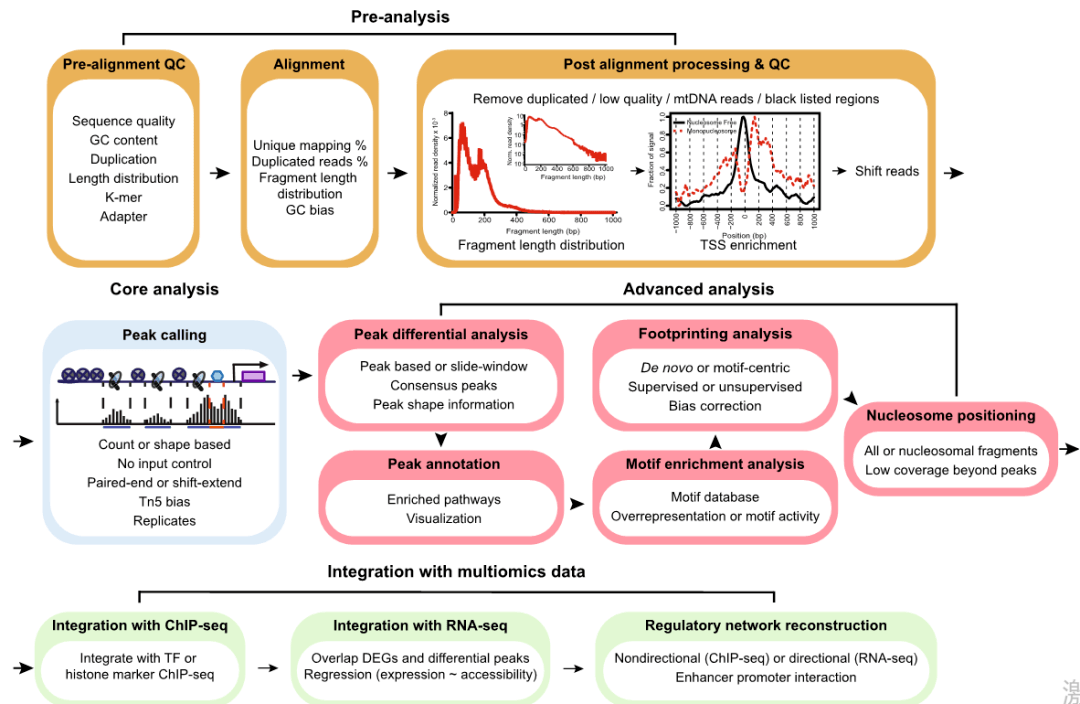
二、预处理(质控和匹配)
这一步包括3个小步骤:(1)比对前QC;(2)read比对;(3)比对后质控和处理。
1.质控:首先比对前需要通过FastQC展示碱基的质量,接下来通过trimmomatic进行去除已知的adapter序列。
2.比对:使用BWA-MEM或者Bowtie2对于短的双端reads进行比对,成功率达到80%以上则认为是比较成功的。对于哺乳类动物,建议最少在开放染色质的区域为5000万个reads,并且至少有200百万TF的足迹。
3.比对后质控:得到比对后的BAM文件后,在线粒体以及ENCODE上的blacklist区域的片段需要被移除(具有极高的read覆盖率),另外因为PCR导致的高重复率同样需要排除。
另外ATAC-seq特有的质控标准同样需要评估,比如片段大小分布图(fragment size distribution)需要梯度式下降,分别是无核小体区(<100bp)、单核小体(200bp)、双核小体(400bp)、三核小体区域(600bp)(Figure 1b)。
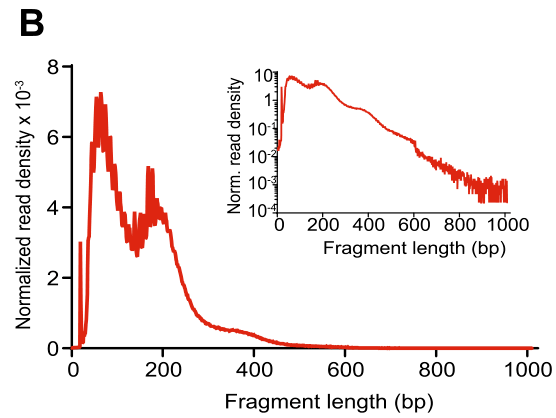
(Figure 1b)
来自于无核小体区域的片段通常在转录起始位点(TSS)区域明显富集,而核小体结合的区域则是在TSS位置信号是缺失的,但是在两侧明显富集(Figure 1C)。这些可以通过ATACseqQC进行评估。
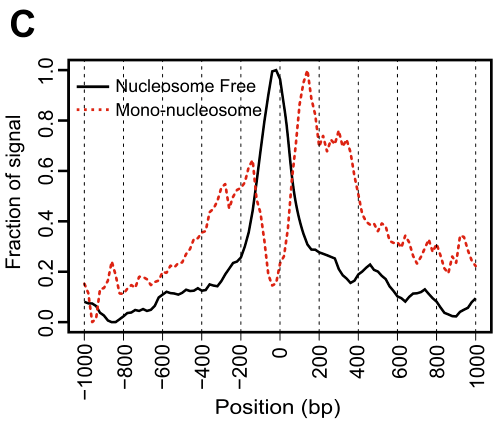
(Figure 1C)
所以标准化的标准流程是:FastQC➔ trimmomatic➔BWA-MEM➔ATACseqQC。
三、Peak鉴定(核心分析)
Peak鉴定在我们前面的推文中也提到过,主要是对peak所映射的染色体位置信息以及对应的基因。类似于Chip-seq或者DNase-seq,ATAC-seq也推荐使用MACS2进行peak call。但是于Chip-seq不同的是,ATAC-seq不需要input作为control组进行对照。并且同时对无核小体区域片段以及核小体结合片段进行peak鉴定(Figure 3A)。并进一步通过平移延伸(shift-extend)的方式来对无核小体区域的peak进行平滑处理(Figure 3B)。
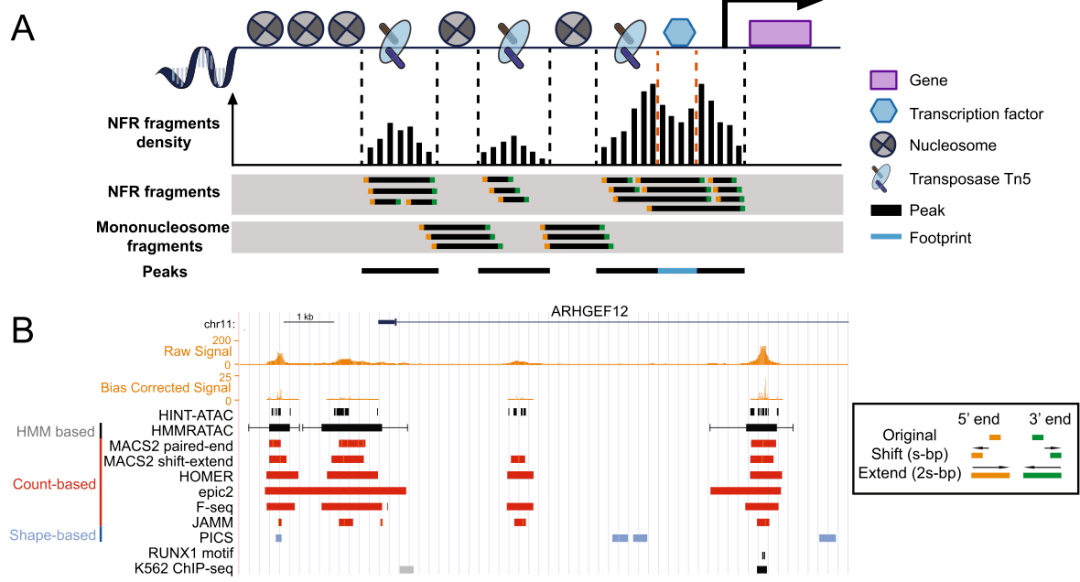
(Figure 3A,B)
目前流行的peak caller主要分为两大类:
1.基于count
2.基于shape 的peak caller。
前者包括MACS2、HOMER等,后者包括PICS、PolyaPeak等。
ATAC-seq主要适用于前者(Figure 4)。其中ATAC专属的HMMRATAC计算效果比MACS2和F-seq效果都要好,并提供了额外的核小体位置信息。
(PS:这里作者推荐一般来说使用MACS2和HOMER进行peak calling。如果计算资源充足,则可以考虑HMMRATAC)
四、ATAc-seq高级分析
peak相关高级分析
1
peak的差异分析
现目前并没有专门为ATAC-seq涉及的差异peak分析工具,目前的差异分析工具根据分析方法同样分为两类,分别是1.consensus peak和2.sliding window-based。对于前者包括HOMER、DBChip、DiffBind依赖于RNA-seq差异分析所需要的包,比如edgeR、DEseq2所作的分析。其中DiffBind或者DBChIP具有交集或者union的选项。后者包括ChIPDiff、PePr等等(Figure 4)。
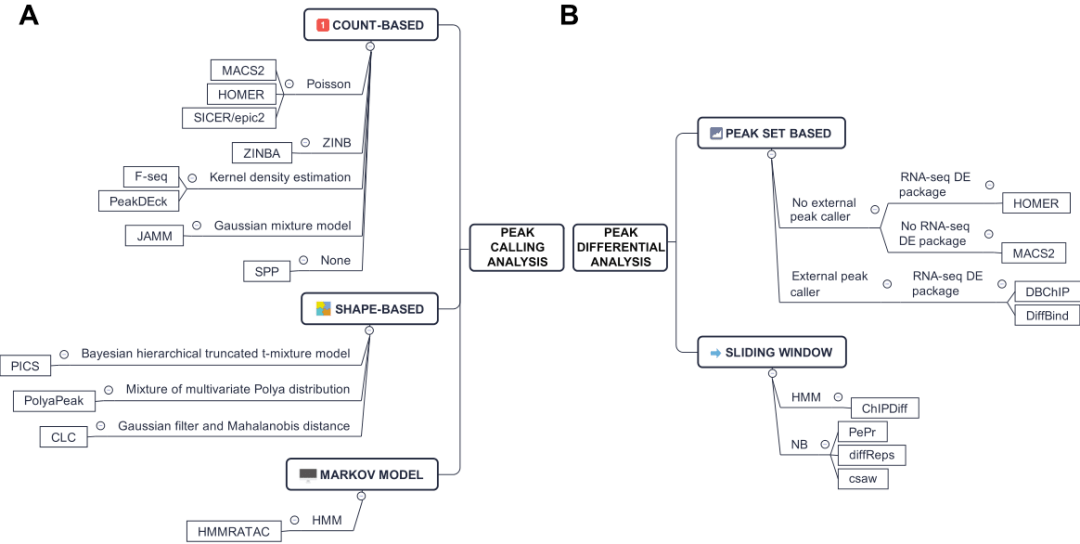
(Figure 4)
2
peak注释
获取到peak的集合之后,注释peak可以使用HOMER、ChIPseeker、ChIPpeakAnno等工具。这些工具能够将peak映射到最近的基因以及对应的基因区域,并且可以通过饼图的方式展示注释的基因特征(Figure 1D),除此之外还可以进行GO、KEGG等功能注释。
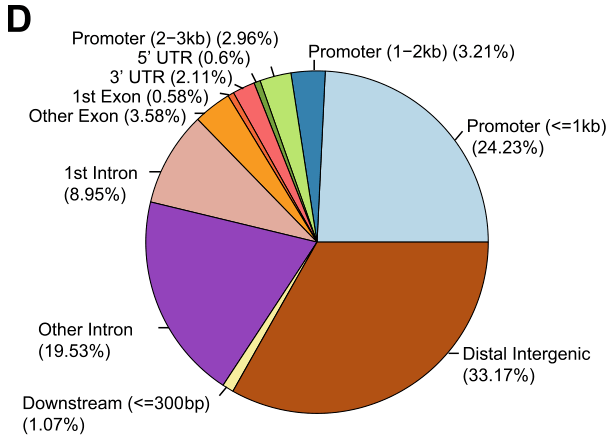
motifs分析
虽然ATAC-seq能够对peak进行注释以及相关的功能富集分析,但是并不能对潜在机制进行解读。而motif则是TF和DNA结合的特定序列,TF所结合的特定位点则是TFBS(TF binding site)。大部分TF结合在染色质开放区域,但是少数的pioneer TF则能够结合到并不完全开放的区域。而转录因子结合的位点能够导致片段在这一区域富集水平相对下调,从而形成TF足迹(footprint)。人类大概有1600多个TF,其中超过一半通过实验和计算获得对应的motif。TF通过和组蛋白以及非组蛋白竞争结合到DNA上发挥转录调控作用。目前有两种对于motif分析的方法:1.基于序列的预测进行motif的频率以及活性预测;2.footprint去计算TF的占有率。
1
motif数据库以及筛选
目前普遍使用的数据库包括JASPAR等数据库,motif以文本的格式保存为PWM的形式。HOMER以及R包TFBSTools、motifmatchr能够通过PWM给定的核苷酸序列搜索对于潜在的TF结合位点。而MEME和PWMScan由于它的网络交互页面所以具有更好的使用体验。
2
motif的富集和活性分析
通过前面的motif搜索工具,每个peak区域的motif的位置和频率信息就有了,接下来可以通过HOMER或者MEME-AME进一步计算TF的出现频次和活性,并和背景信号进行比较,从而推测转录因子的活性。除此之外,还包括ChromVAR能够计算每个motif在多个分组中的可及性,并用Z score的方式进行评分(这是专门为scATAC-seq数据设计的分析)。
3
TF足迹(footprint)分析
另一个评估TF调控的方式则是footprint。Footprint是因为TF结合在DNA上阻碍了Tn5酶的剪切,从而留下了开放染色质区域种的一个相对缺失(波谷)。但是目前对ATAC-seq的分析是存在障碍的:(1)首先在预处理的时候需要移动原始read,由于具有 9个碱基的重复;(2)由于Tn5的亲和力很强,并且TF短暂结合具有较弱的结合能力。所以footprint并不容易检测。因此footprint检测并不准确。
目前分析footprint的工具包含两类:(1)de novo以及(2)motif-centric方法(Table 1)。前者基于footprint的特点模式(peak-dip-peak)进行预测,接下来则通过推定的footprint位点与已知的motif进行联系或者是验证发现新的motif。后者则是基于先验的TFBS并且通过监督以及无监督的方式区别这些位点是否是结合的。De novo tools方法包括HINT-ATAC校正了链特异性的Tn5剪切偏倚。Motif-centric tools这类方法作者推荐BaGFoot工具,能够计算footprint depth并且对测序深度以及偏倚进行校正。总地来说作者推荐HINT-ATAC工具。
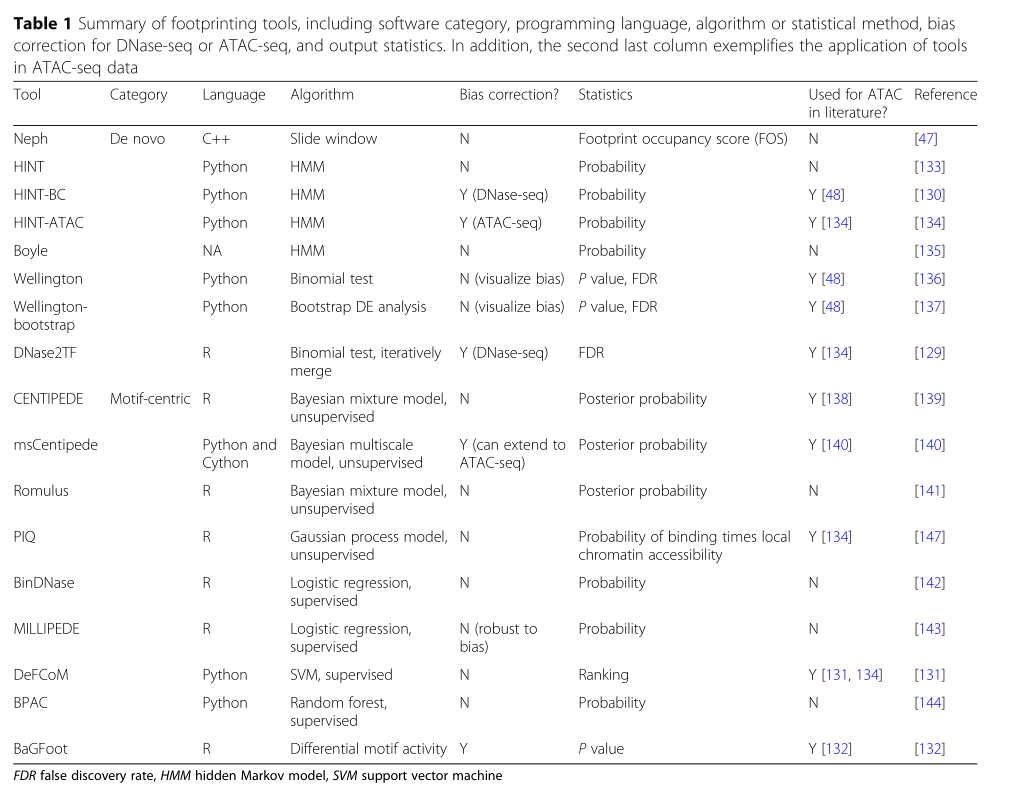
核小体位置分析
核小体由组蛋白形成的八聚体以及147bp的DNA构成。在ATAC-seq分析种,更长的DNA片段通常是由于核小体相关的区域导致的。但是检测出核小体的覆盖率要低于MNase-seq。HMMRATAC and NucleoATAC是两个最常用的ATAC-seq核小体检测的方法。
五、多组学数据整合分析构建调控网络
1
ChIP-seq数据整合
由于开放染色质同上是TF结合的前提,所以ATAC-seq peak通常与TF的ChIP-seq进行重叠,但是前者的范围更广。所以两者可以相互验证。只在TF的ChIP-seq中存在的peak才可以视作为pioneer TF结合到关闭的染色质范围上。对于ATAC-seq的motif和TF足迹分析能够进一步整合到真实的TF的ChIP-seq中,从而降低假阳性率。同样ATAC-seq能够和组蛋白的ChIP-seq进行分析,并找到组蛋白对于染色质的开放促进还是抑制作用(与开放区域转录正相关,如H3K4me3, H3K4me1, H3K27ac等,以及与开放区域转录负相关,如H3K27me3)
2
RNA-seq整合
差异基因同能能够逆向推定上游的TF,并找到在开放染色质特定的footprint和motif。通常是scATAC-seq和scRNA-seq的联合。
3
构建regulatory网络
这里最常使用在scATAC-seq分析,比如Cicero包进行的STAC-se增强子-基因调控网络(Figue. 3C)。
好啦,关于ATAC-seq的综述就讲解到这里啦,我是风间琉璃,我们下期见~
- 本文固定链接: https://maimengkong.com/kyjc/1308.html
- 转载请注明: : 萌小白 2022年12月15日 于 卖萌控的博客 发表
- 百度已收录
