原理
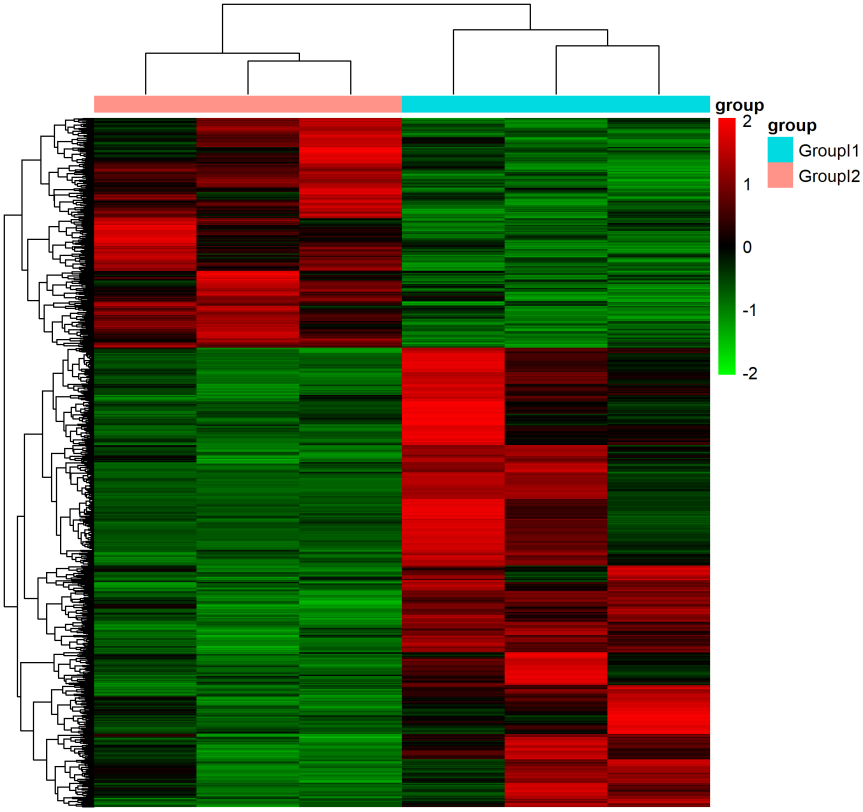
提起热图,大家可能马上就能领会:热图不就是差异表达基因在不同分组样品中的表达水平吗?一般一列表示一个样品,一行表示一个基因,其中的颜色表示的相对表达值。但说到热图与机器学习,很多人都会惊讶:一个简单的热图与高大上的机器学习能扯上什么关系?
都说“越熟悉越陌生”,这话放这一点儿不假;大部分人都接触或亲手绘制过热图,但在这个“过度包装”的时代(很多软件和在线工具都实现了“一键生成”),我们都选择性忽略了热图其实是聚类的可视化结果。
“物以类聚,人以群分”,人一出生就被教育给世界打上标签:最初的“爸爸/妈妈”与?.?,然后是“好人”与“坏人”,接着是“自己人”与“其他人”,……在这里,形成了最雏形的概念:特征与分类。那些数学家整天也没闲着,总想着“一生二,二生三,三生万物”以及“将具体抽象化”(总想着“找茬”),最后将分类拓展成了机器学习最核心的一大分支。当然,分类问题根据数据特征又被细化许多不同的类别,而热图只涉及了一小部分 —— 层次聚类。
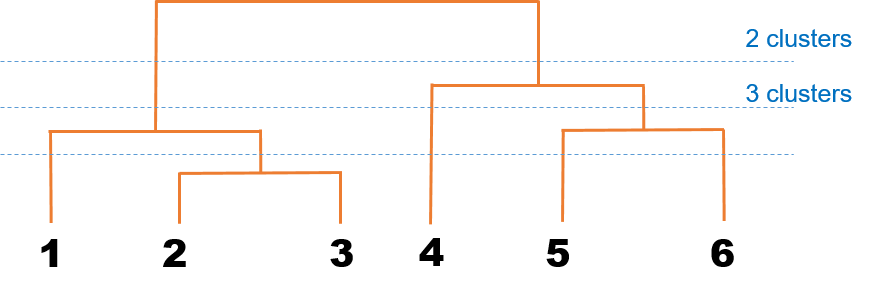
实际上,热图的顶部面板的树状图与左侧面板密密麻麻的树状图展示的就是层次聚类的基本思路:计算两个对象之间的距离,然后根据距离划分类别(如图)。
可以看出层次聚类相对简单,关键是计算对象之间的距离,计算距离常用方法包括:
- euclidian 欧式距离
- correlation 相关性
- manhattan 曼哈顿距离
- minkowski 名科夫斯基距离
- maximum 最大距离
- …
计算完距离之后,我们需要根据距离将不同对象汇集在一起,即:第一次 2 与 3 聚为一类,其他对象各自为政;第二次 1/2/3 为一类而 5/6 为一类,4 单独为一类;第三次 1/2/3 与 4/5/6 分为两类。但怎样聚类,比如在第二次分类时,我们会遇到一个问题:1 与 2/3 距离怎么计算?下面是解决方案:
- single linkage 计算两个类中的最小距离
- average linkage 计算两个类点与点距离的平均
- complete linkage 计算两个类中的最远距离
- centroid linkage 计算两个类质心的距离
- ……
当然,层次聚类的具体过程比我所描述的更复杂一点儿。
用 R 实现聚类
在 R 中,基础包 stats 提供了一个层次聚类的函数hclust,即 Hierarchical Clustering 的缩写。该函数基本思路是:
- 将需要聚类的对象单独作为一类 cluster
- 根据算法(比如 complete linkage)将两个最相似的 cluster 聚为一类
- 重复 2 过程,直到所有 cluster 都聚为一类
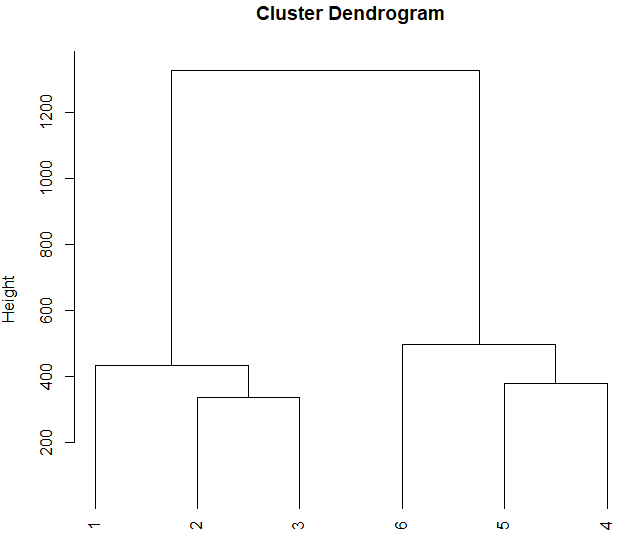
现在我们有 6 个样品的差异基因表达谱exp_data,现在想对样品进行聚类:
# exp_data 是一个矩阵:行为基因,列为样品,值为表达值。这里不展示
# 根据表达值计算 6 个样品的距离
sm_dist <- dist(t(exp))
# 显示距离矩阵,可以看出是一个 下三角矩阵
sm_dist
1 2 3 5 6
2 1561.485
3 2594.585 2058.536
5 11914.911 12963.577 12980.751
6 5649.365 6601.354 6166.485 11180.593
4 22901.096 23918.870 23877.047 11556.278 21616.226
# 利用 hclust 进行聚类
hc <- hclust(sm_dist)
# 显示结果,可以看到聚类方法以及计算距离的方法
hc
Call:
hclust(d = sm_dist)
Cluster method : complete
Distance : euclidean
Number of objects: 6
# 可视化
plot(hc, hang = -1)
常用工具
从基因芯片到二代测序再到最新的三代、四代测序,热图的绘图工具也经历了时间的洗礼。
- stats::heatmap 最初也是最基本的(还记得最初的配色吗?)
-
- pheatmap 应该是使用频率最高的工具
- gplot::heatmap.2 使用频率也非常高
- ggplot2 自己从头到尾操作一遍印象深刻
此热图非彼热图
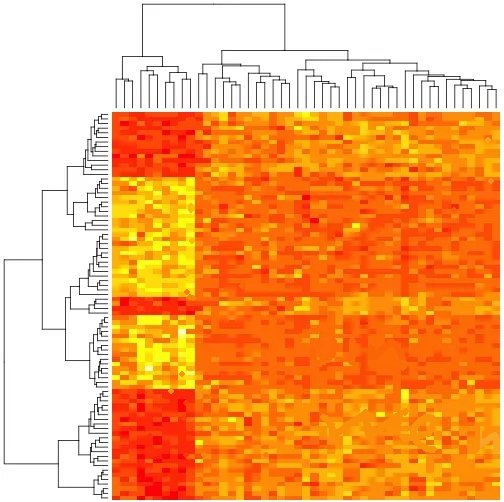
除了基因表达谱的热图,相信大家都见过这样的图
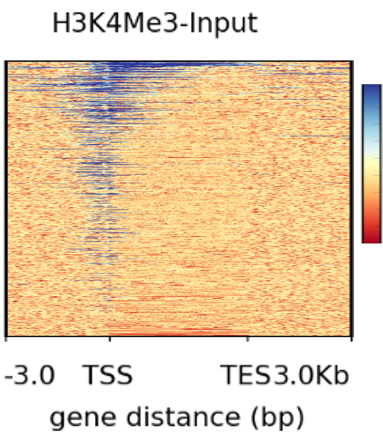
实际上,这也是一种热图,常见于 ChIP-seq,被称为 tag density heatmap。
与 RNA-seq 不同,ChIP-seq 或 CLIP-seq 通常人们把落在基因组上的 reads 称为 tag。
可以看出,表达热图与基因区域热图似乎完全不一样,那基因区域热图展示的到底是什么呢?
首先我们看一下该图的结构:
热图的本质仍是矩阵
- 横坐标
- 基因区域,转录起始位点的上游 3kb 到转录终止位点下游的 3kb。实际上,该区域被分割成许多小的区间 bin,例如每个 bin 长度为 25bp。
- 纵坐标
- 代表的是基因,一般来说表示有 reads 覆盖的基因
- 颜色与值
- 由横纵坐标我们可以看出,每个小格表示一个基因在一个 bin 的表达信号强度。
- 聚类
- 将表达相似的行聚在一起。
所以,这幅图展示给我们的是:H3K4me3 结合的 tags 主要富集到了转录起始位点 TSS 的上下游区域。
总的来说,不管是基因表达热图还是这里的热图,其数据结构都是矩阵,不过 tag density heatmap 展示的是基因组区域,由于行与列很多(目前已知基因约 2.5 万个;每个 bin 只有 25bp,那么宽度为 10kb 的区域将被分割成 400 份)所以显得整幅图密密麻麻。
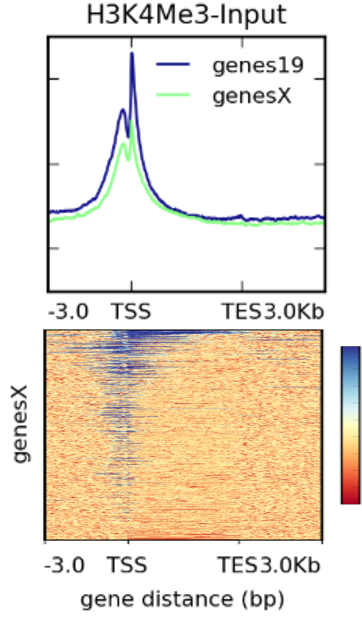
由于 heatmap 图整体过于庞大,一般人们会选择更为直观的 profile plot(即热图上面的曲线图)。
- 本文固定链接: https://maimengkong.com/kyjc/1194.html
- 转载请注明: : 萌小白 2022年10月1日 于 卖萌控的博客 发表
- 百度已收录
