前面已经讲述了R包用clusterProfiler做GO富集分析clusterProfiler的GO富集分析方法,本篇继续演示R包goseq的GO富集分析。
相比clusterProfiler中的GO富集分析方法,goseq的特别之处在于,不再使用超几何分布(Hyper-geometric distribution)检验,而是使用了Wallenius non-central hyper-geometric分布。除了背景基因的数量外,它还同时考虑了基因长度信息,认为从某个类别中抽取个体的概率与从某个类别之外抽取一个个体的概率是不同的,这种概率的不同是通过对基因长度的偏好性进行估计得到的,从而其认为能更为准确地计算出 GO term被差异基因富集的概率。
goseq包的安装
对于goseq的安装也很简单,一般情况下,直接通过Bioconductor安装goseq就可以了。
!!!**************************************************
#bioconductor 安装
#install.packages('BiocManager') #需要首先安装 BiocManager,如果尚未安装请先执行该步
BiocManager::install('goseq')
!!!**************************************************
goseq的GO富集分析(有参向)
这里均对于有参考基因组的情况而言的,无参分析暂不涉及。
就以人类转录组数据为例展示GO分析的过程吧。
1、准备输入数据
需要准备两类数据。
(1)待分析的基因名称,例如这里以人类参考基因组hg38版本的ensembl id为例。把基因名称以一列的形式排开,放在一个文本文件中(例如命名“gene_select.txt”)。Excel中查看,就是如下示例这种样式。
 输入数据,基因名称李列表
输入数据,基因名称李列表
(2)所有基因长度信息,既包含待分析的基因,也要包含背景基因,根据所选择的参考基因组定义。例如这里统计了人类参考基因组hg38版本的所有基因长度,第一列是基因名称,第二列是基因长度,放在一个文本文件中(例如命名“gene_length.all.txt”)。Excel中查看,就是如下示例这种样式。
 输入数据,基因长度信息
输入数据,基因长度信息
2、goseq的GO富集分析
首先读取基因列表和长度文件,预处理数据。
!!!**************************************************
#读取待GO分析的基因名称,该示例来自人参考基因组 hg38 的基因名称
de_gene <- as.vector(read.delim('gene_select.txt', sep = '\t')[[1]])
#读取人类 hg38 的所有基因长度信息
gene_list <- read.delim('gene_length.all.txt', sep = '\t', row.names = 1)
#设置待富集的基因为 1,背景基因为 0
genes <- rep(0, nrow(gene_list))
names(genes) <- rownames(gene_list)
genes[de_gene] <- 1
head(genes) #处理后的待分析数据
!!!**************************************************
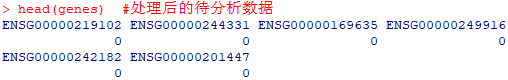
结果获得一组向量结构在R中存储。向量中记录了所有基因的名称,以及其是作为背景基因存在(0)还是作为富集基因存在(1)。
随后加载包goseq,并使用goseq的内部函数enrichGO()即可完成GO富集分析。
!!!**************************************************
library(goseq)
#根据基因长度加权
pwf <- nullp(DEgenes = genes, bias.data = gene_list$gene_length, plot.fit = FALSE)
head(pwf)
#GO 富集分析
#hg38 是内置的人类 hg38 基因组参考库
#ensGene 意为基因名称使用 ensembl id 作匹配进行富集
pvals <- goseq(pwf, 'hg38', 'ensGene')
head(pvals)
#手动对 P 值作个校正,例如 FDR 校正
pvals$FDR <- p.adjust(pvals$over_represented_pvalue, method = 'fdr')
pvals <- pvals[c(1,2,8,4,5,6,7)] #第三列的p值就不要了
head(pvals)
!!!**************************************************
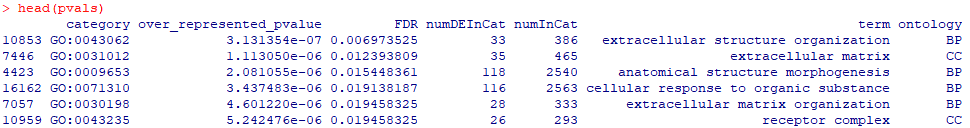 goseq的GO富集分析结果表格
goseq的GO富集分析结果表格
对于各列内容:
category,富集到的GO id;
over_represented_pvalue,富集的p值;
FDR,p调整值;
numDEInCat和numInCat,分别为富集到该GO条目中的基因数目,以及该条目中背景基因总数目;
term,富集到的GO的描述信息;
ontology,GO分类BP(生物学过程)、CC(细胞组分)或MF(分子功能)。
3、手动添加基因名称
我们看到,上述goseq的默认输出中,只统计了富集到该GO条目中的基因数量,并未把具体的基因名称展示出来。如果需要,基因名称我们来手动匹配下。
!!!**************************************************
#添加基因,参考方案
#https://www.biostars.org/p/355247/
getGeneLists <- function(pwf, goterms, genome, ids){
gene2cat <- getgo(rownames(pwf), genome, ids)
cat2gene <- split(rep(names(gene2cat), sapply(gene2cat, length)),
unlist(gene2cat, use.names = FALSE))
out <- list()
for(term in goterms){
tmp <- pwf[cat2gene[[term]],]
tmp <- rownames(tmp[tmp$DEgenes > 0, ])
out[[term]] <- tmp
}
out
}
goterms <- pvals$category
goList <- getGeneLists(pwf, goterms, 'hg38', 'ensGene')
#默认将富集到GO的基因名称添加在最后一列,然后输出到本地
pvals$Ensembl_ID <- sapply(pvals$category, function(x) paste(goList[[x]], collapse = '; '))
write.table(pvals, 'goseq.txt', sep = '\t', row.names = FALSE, quote = FALSE)
!!!**************************************************
 添加了带富集基因名称的输出表格
添加了带富集基因名称的输出表格
内容格式见上文,最后一列添加了富集到该条目的基因名称信息。
由于输入文件使用的ensembl id,因此最后展示的也是ensembl id。如果期望使用其它的基因名称,如通俗的symbol id等类型,除了更改输入文件为使用symbol id的基因名称做分析外,还可以通过基因名称转换的方式对ensembl id和symbol id作个匹配转换。
!
!
!
注:!!!*******之间为R脚本内容。
搜索微信公众号“纪伟讲测序”- 本文固定链接: https://maimengkong.com/kyjc/1010.html
- 转载请注明: : 萌小白 2022年6月23日 于 卖萌控的博客 发表
- 百度已收录
