使用Perseus软件进行1D和2D annotation enrichment分析
某天,小编在某篇蛋白组学文献中看到这么一个分析,称为2D annotation enrichment的方法。作者使用2D annotation enrichment比较了自己的蛋白组数据集和先前研究中蛋白组数据集的蛋白表达的差异,并将这种差异关联到富集的通路上进行比较,以阐述二者的一致性和区别。图B是2D annotation enrichment的结果,横轴为自己的蛋白组数据,纵轴为先前报道的蛋白组数据,横轴和纵轴坐标表示了这些通路在两个数据集中的富集因子,正值代表激活,负值代表抑制;图中的点代表了富集的通路,不同颜色的点代表了不用类型的功能通路。这样,功能通路在两个数据集中的状态清晰可对比,看到相似的功能通路在两数据集中具有相似的激活或抑制特征。
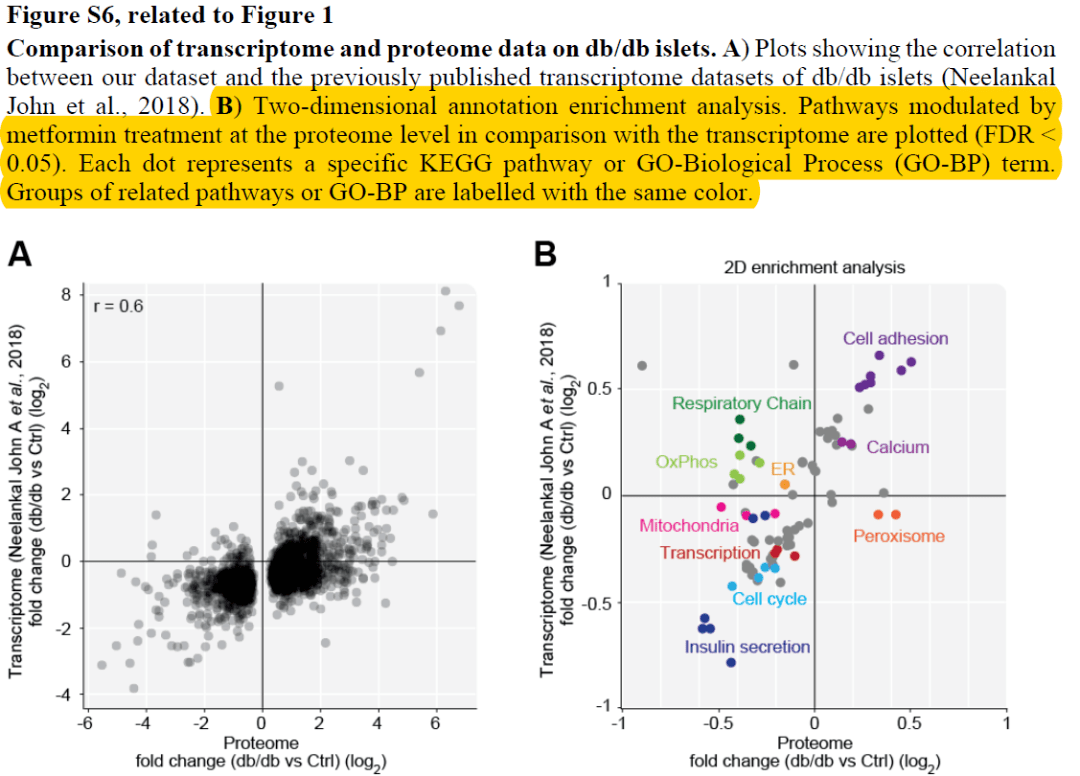
来源文献,Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets
令小编好奇的是,这个2D annotation enrichment分析是如何根据两组数据集中蛋白水平的倍数变化,关联到通路水平进行比较,解释生物学问题的呢?本篇就让我们重现这种分析方法。
2D annotation enrichment可以对两个不同的组学数据进行联合分析,以对比它们的一致性与不同。例如,对蛋白组和转录组数据集进行联合分析,根据所有蛋白或基因表达的倍数变化值,计算对应的通路是否受到激活或抑制。根据结果,可以用于描述哪些通路在转录组水平激活而翻译水平未体现,或者在翻译水平激活但转录水平未体现,以阐述转录水平和翻译水平基因表达的生物学现象。或者,根据一致的通路描述二者的共性,描述功能的一致性。
目前,2D annotation enrichment可以使用Perseus软件来完成分析,Perseus下载地址:https://www.maxquant.org/perseus/
这应该是关于2D annotation enrichment的第一篇中文教程了。为了方便大家操作,测试数据,作图R代码等,可在微信公众号阅读原文获取。
1 输入数据格式
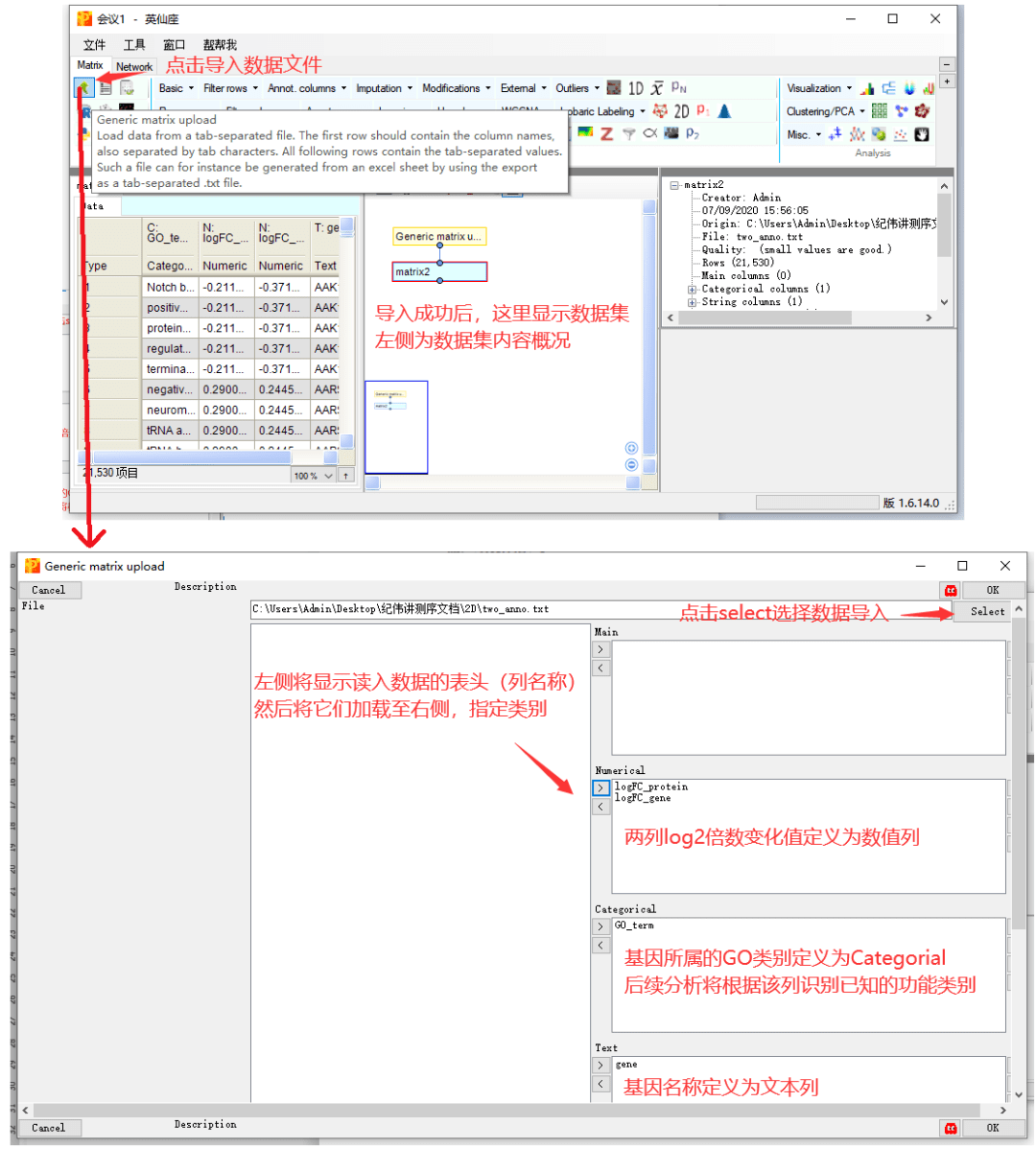
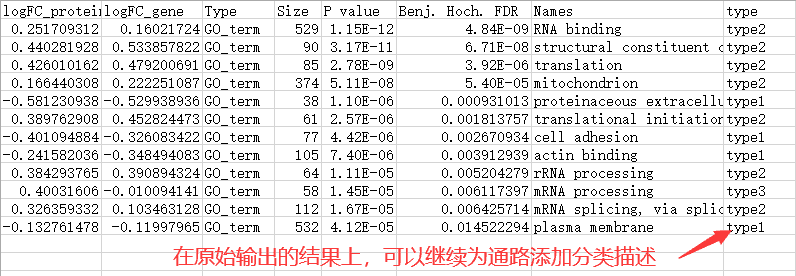
输入数据文件以制表符分割,包含四列信息,记录了基因名称,转录组和蛋白组中计算的基因的差异表达倍数,以及基因所属的功能类别。
以该示例数据为例:
gene,基因名称列,记录基因的名称,可以为任意名称。
logFC_gene和logFC_protein,log2转换后的差异倍数,一列是基因对应的蛋白的差异表达,一列是基因对应的转录组的差异表达。
GO_term,基因所属的功能条目,该示例中展示了基因归类的GO功能类别。
由于一个基因可以归属多条功能,因此基因名称或者功能条目均可以出现多次。
2 数据导入至Perseus
Perseus的安装过程就不再多说了,很简单,下载就能用。界面也是图形化界面,很好操作。现在我们打开Perseus,将准备的数据读取到Perseus中用作功能富集分析。
软件左上方有个导入按钮,点击后在新界面选择本地的文件读取,并设置列的类型后,点击确定即可成功读入。
3 1D annotation enrichment
接下来就是令人激动的分析环节了,我们首先来看单组学数据集的功能分析过程吧,也就是1D annotation enrichment。
例如这里以蛋白组数据集为例,根据所有蛋白的倍数变化值,计算这些蛋白对应的通路是否受到激活或抑制。
备注:就目前而言,1D annotation enrichment很少使用,更多地使用基因集富集分析(GSEA)实现类似的目的。
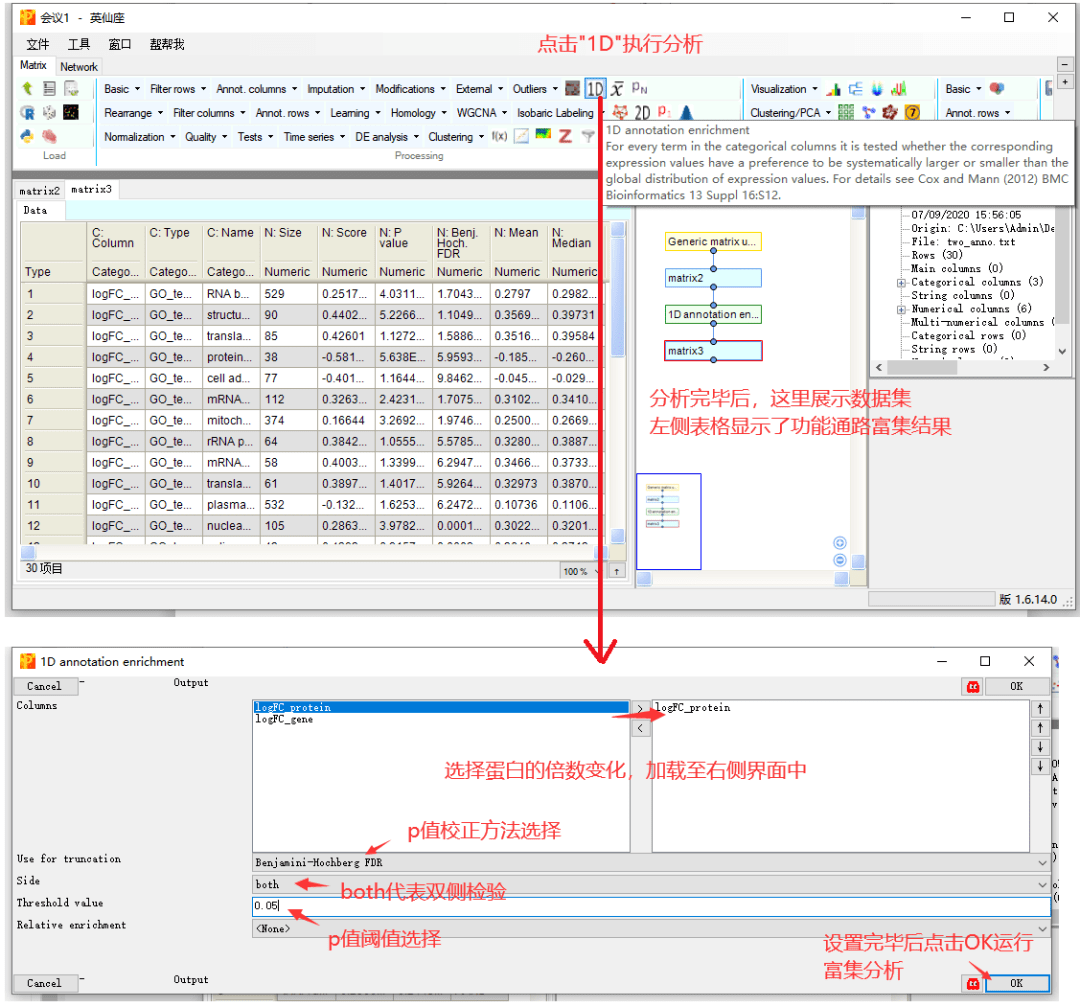
结果表格中,重点关注几列信息就可以了。
C:Name,为富集的功能条目名称,本示例中,为基因所属的GO功能类别的名称。
N:Size,富集到该功能的蛋白数量。
N:Score,富集得分,取值范围-1~1,正值代表了该通路的激活,负值代表了该通路的抑制。
N:Pvalue和FDR,分别为通路富集的p值和p调整值,只保留了设定在阈值范围内的显著结果。
对于富集结果的输出,点击右上方的保存按钮,输出在本地文件保存。
4 2D annotation enrichment
2D annotation enrichment分析是Perseus的特色,可以对两个不同的组学数据进行联合分析,以对比它们的一致性与不同。
接下来,我们同时选择蛋白组和转录组数据集,根据所有蛋白或基因表达的倍数变化值,计算对应的通路是否受到激活或抑制。根据结果,可以用于描述哪些通路在转录组水平激活而翻译水平未体现,或者在翻译水平激活但转录水平未体现,以阐述转录水平和翻译水平基因表达的生物学现象。或者,根据一致的通路描述二者的共性。
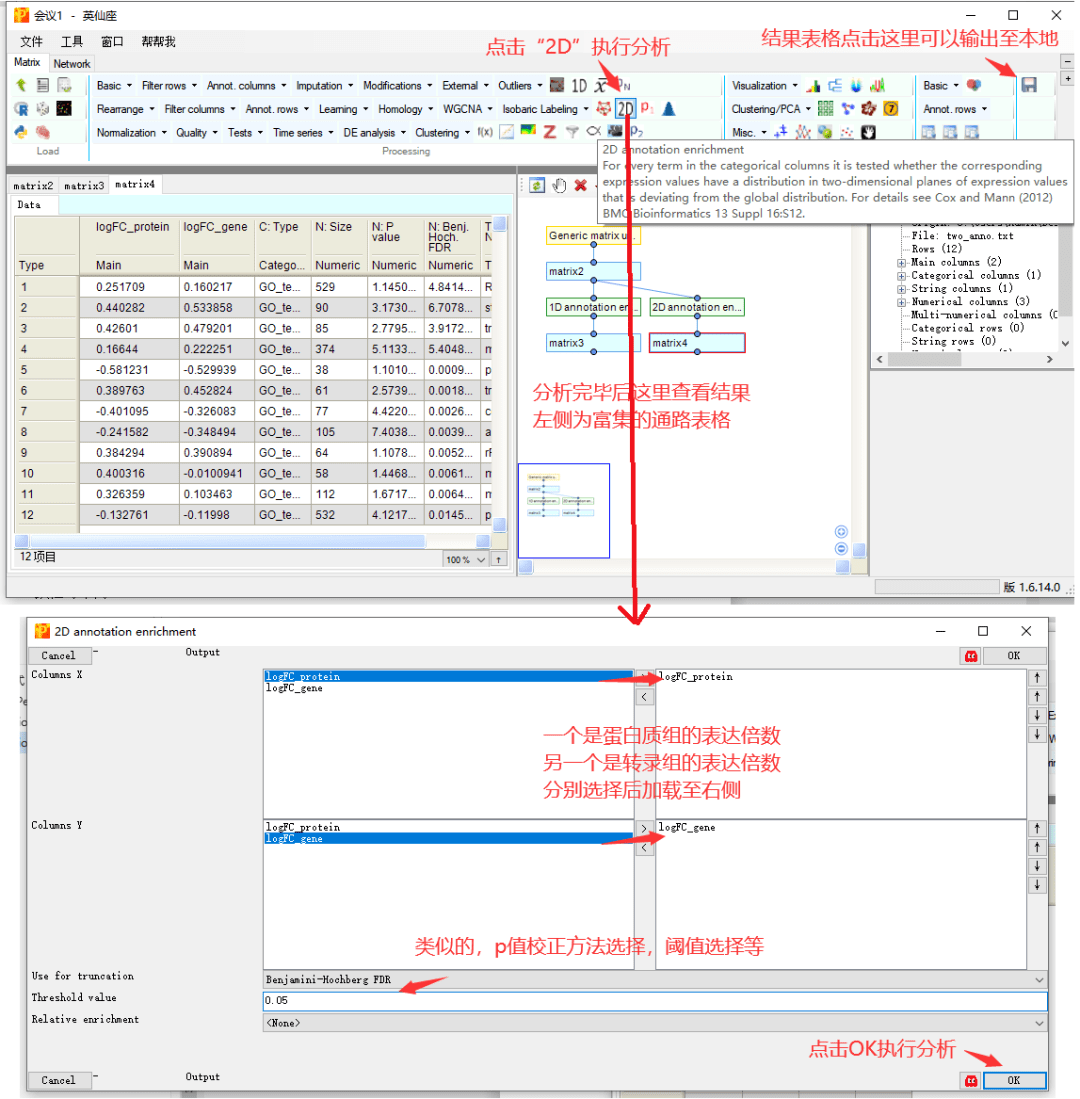
类似地,结果表格中,重点关注几列信息就可以了。
logFC_protein和logFC_gene,分别为蛋白和转录水平的富集得分,取值范围-1~1,正值代表了该通路的激活,负值代表了该通路的抑制。
T:Names,为富集的功能条目名称,本示例中,为基因所属的GO功能类别的名称。
N:Size,富集到该功能的基因数量。
N:Pvalue和FDR,分别为通路富集的p值和p调整值,只保留了设定在阈值范围内的显著结果。
对于富集结果的输出,点击右上方的保存按钮,输出在本地文件保存。
5 R语言的可视化
通路的富集分析结果拿到手了,最后就是作图展示。
结果输出后,稍作处理,例如可以继续为功能添加分类。对于分类的添加,可以是进一步的功能大类,将通路关联到更大的分类中,这样更有利于描述功能的一致性。或者,根据蛋白组和转录组的通路富集的一致性趋势划分分类也可以。这里直接按四象限分布划分分类了,仅用作示例展示。
最后,我们使用R语言ggplot2包绘制二维散点图表示通路富集,就以2D annotation enrichment 得到的蛋白组和转录组双组学的联合分析为例展示。
!!!********************************************************************
#2D使用Perseus计算
#并手动添加功能分类后,在R里作图
dat <- read.delim('2D_result.txt')
#散点图
library(ggplot2)
library(ggrepel)
ggplot(dat, aes(logFC_protein, logFC_gene)) +
geom_point(aes(color = type), size = 2) +
theme(panel.grid.minor = element_blank(), axis.line = element_line(color = 'black')) +
geom_vline(xintercept = 0, linetype = 2) +
geom_hline(yintercept = 0, linetype = 2) +
labs(x = 'Proteome\nlog2FC', y = 'Transcriptome\nlog2FC', color = '') +
scale_x_continuous(expand = c(0, 0), limit = c(-1, 1)) +
scale_y_continuous(expand = c(0, 0), limit = c(-1, 1)) +
geom_text_repel(aes(label = Names), size = 3,
box.padding = unit(0.5, 'lines'), show.legend = FALSE)
!!!********************************************************************
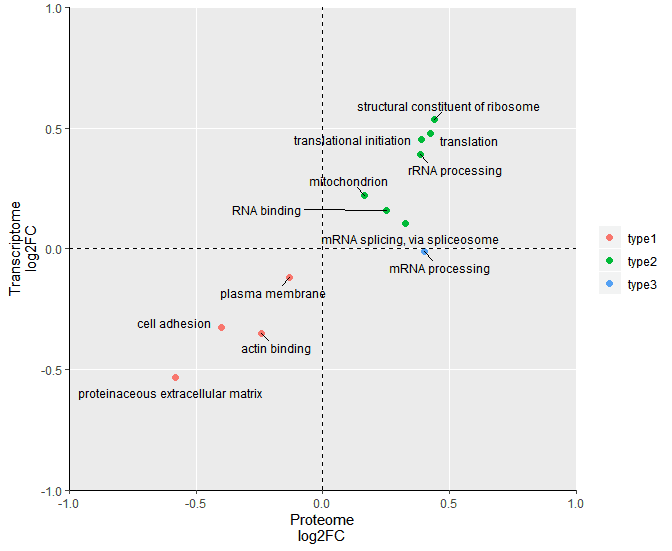
这样,成品图就出来了,后续根据两个组学数据富集通路的一致性或区别,描述生物学现象就可以了。
纪伟点评:我们经常听到多组学分析,多数据集关联分析,这些联合分析可以很好的说明分子及功能的变化规律的普遍性,稳定性,但联合分析出图是个大问题。
分子水平差异变化:如转录组mRNA, lncRNA, circRNA, miRNA与蛋白,磷酸化蛋白,代谢小分子,基因突变,染色体变异等;及不同来源数据集(公共数据)分子的比较如何出图。
功能水平上:富集重要信号通路如增殖,粘附,胰岛素分泌等关心的通路如何在多组学活多数据集上展示差异及共性,如何出图。
信号轴上:某个具体的信号轴上,多组学的分子,不同的数据集如何展示其差异与共性,揭示处理条件下特有的信号轴变化,如何出图。
本篇文章中的分析,给我们解决功能富集关联提出了一个很好的方法,首次看到它时,着实让我兴奋不已,此刻依然心潮澎湃...
-------------------------------------------------------------------------
!
!
!
注:!!!*******之间为R脚本内容。
- 本文固定链接: https://maimengkong.com/kyjc/1054.html
- 转载请注明: : 萌小白 2022年6月28日 于 卖萌控的博客 发表
- 百度已收录
