这次我们来个GC含量的圈图,同样的,我在这里也会描绘出各种GC含量圈图的画法,开开眼界。GC含量图在基因组circos图中算是非常有分量的,各种GC含量圈图的表达形式6的飞起。下面开始画几种类型的GC圈图。
首先,加载circlize R包
library(circlize)
读入染色体数据和GC含量数据
data<-read.table("chrom.txt",head=T,stringsAsFactors=FALSE,sep='t')
circos.genomicInitialize(data,plotType="NULL")
circos.track(ylim = c(0, 1), panel.fun = function(x, y) {
chr = CELL_META$sector.index
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.rect(xlim[1], 0, xlim[2], 1, col = rand_color(14))
circos.text(mean(xlim), mean(ylim), chr, cex = 1, col = "white",
facing = "inside", niceFacing = TRUE)
}, track.height = 0.1, bg.border = NA)
这些代码之前的一篇文章我已经提过代码了,所以如果不懂,请参考我前面写的文章
读入GC含量数据:
bed1<-read.table("GC_config.txt",head=T,sep='t')
数据格式如下:
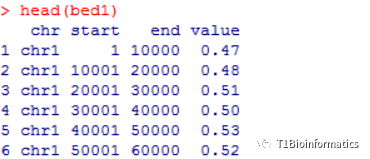
这里GC含量都是采用滑窗进行计算,可以自己写代码也可以用一行命令解决,或者用bedtools。
下面开始画图吧:
类型一:折线GC图
circos.genomicTrackPlotRegion(bed1,panel.fun = function(region, value, ...){
circos.genomicLines(region, value, type = "l",col='green',...)})
##这里面采用的线性类型是I,线的颜色的是绿色
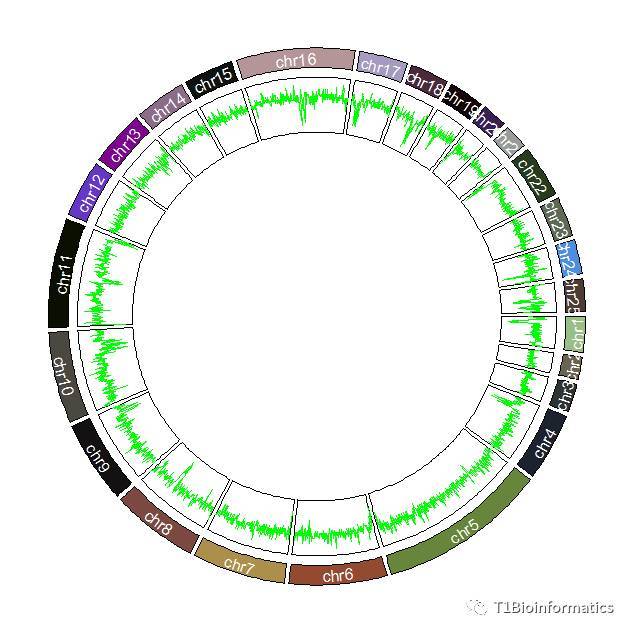
折线图能看出来的是GC含量变化的高低,会有明显的高低峰的区别
类型二:GC含量圈图
前面步骤都一样,我就直接说点图画法:
circos.genomicTrackPlotRegion(bed1,
panel.fun = function(region, value, ...) {
cex = (value[[1]] - min(value[[1]]))/(max(value[[1]]) - min(value[[1]]))#
i = getI(...)
circos.genomicPoints(region, value, cex = cex, pch = 16, col = "red", ...)})
##这里面可以不用管fuction()啥的,知道是个规定就好了,点的大小,我用cex代替,cex是一个变化值,pch选的16,颜色是红色

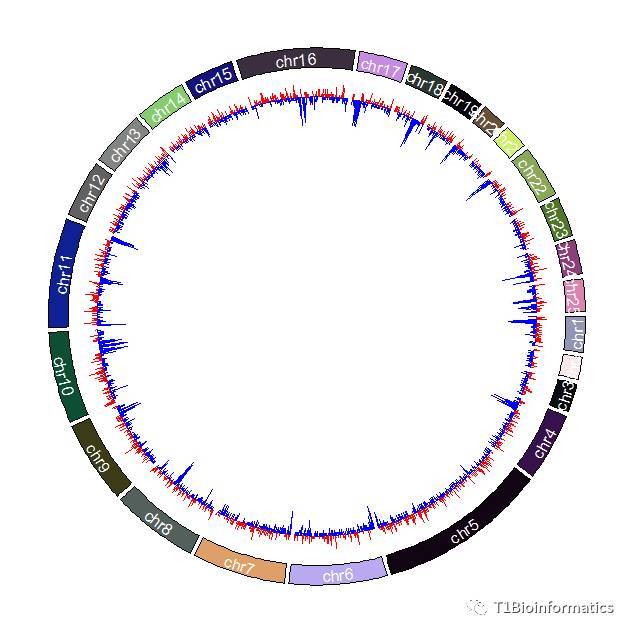
类型三:GC含量热图
f=colorRamp2(breaks=c(0.38,0.46,0.58),color=c("green","black","red"))
circos.genomicTrackPlotRegion(bed1, stack = TRUE,
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = f(value[[1]]), border = NA, ...)
},bg.border=NA)
##这里需要设置颜色棒,bg.border=NA,是将外围的框框去掉,不用框框

可能是我个人审美有问题,颜色配置的丑,别见怪哈,(内心独白就是:你行你上啊),今天就只介绍了这三种GC含量热图,还有好几种,你们可以关注我啊,然后问我,我就给你们代码啊,比如这种以基线为准的热图,大于多少标为正,小于多少向下:
今天还将会展示俩种共线图,觉得不错,可以转载啊。载入R包
library(circlize)
准备数据
我自己造了4个数据,就是为了展示这些图
第一个数据,染色体长度,随机造了6条染色体:

第二个数据,共线性位置信息

第三个数据,对应的共线性位置信息
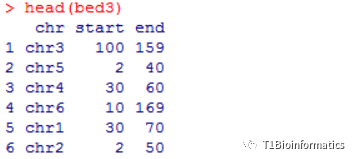
先画第一个共线性图,第四种数据,我待会说
circos.par(gap.degree=5,start.degree=80)
circos.genomicInitialize(bed1)
circos.genomicTrackPlotRegion(bed1,ylim=c(-1,1),track.height=0.05
,bg.col=rand_color(nrow(bed1)))
circos.genomicLink(bed2, bed3,col = rand_color(nrow(bed3)
,transparency = 0.1))
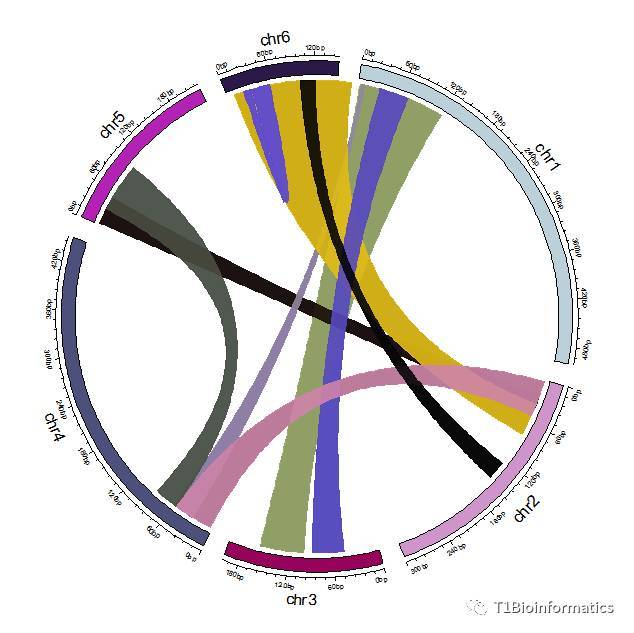
这种图比较适合共线块大,并且共线数据少的类型,但是往往我们利用共线软件获得的共线数据非常多,而且密集,对于这种,我推荐下面一种画法。
配置颜色,相邻位置,或者共线性多的数据的染色体最好配置同种颜色,这里我配置的第四种数据是相邻染色体的共线性多
col1 = c("seagreen","seagreen","salmon","salmon","skyblue","skyblue")
circos.genomicInitialize(bed1)
circos.genomicTrackPlotRegion(bed1,ylim=c(-1,1),track.height=0.05
,bg.col=col1)
names(col1) = unique(bed4[,1])
for(i in seq_len(nrow(bed4))) {
circos.link(bed4[i,1], c(bed4[i,2],bed4[i,3]),
bed4[i,4], c(bed4[i,5],bed4[i,6]),
col =col1[bed4[i,4]])}

鉴于我的审美有限,无法给你们描绘出五彩斑斓的黑色,只能到这样了,代码看不懂的可以看看我之前写的文章。开学了,加油咯!!
参考文献:Zuguang Gu, Lei Gu, Roland Eils, Matthias Schlesner, Benedikt Brors, circlize Implements and enhances circular visualization in R. Bioinformatics
x下面算是circos图的续篇,属于circos的一部分,目前我还没想到这图可以用到哪些生物学数据,但是RNAseq的差异分析应该是可以的。
首先,载入circlize R包
library(circlize)
其次,构建数据,输入的数据为一个矩阵关系。假设我们有A,B,C,D,E 5个组织,分别在正常和干旱产生了若干差异基因,那我们可以构建个矩阵:
data<-matrix(sample (100,100),5,5) ##随机构造5行5列的数据
rownames(data) = c("A","B","C","D","E") ##构建行名
colnames(data) = c("A","B","C","D","E") ##列名
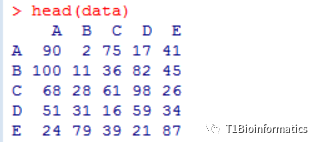
这里我要说明的是,我们A,B,C,D,E五个组织,一个正常和一个干旱,A对A,表示的是A组织在干旱和正常条件差异基因90个,A组织对B组织100个差异基因,这表示的是一个交集,就是A组织的差异基因和B组织的差异基因的交集。这里数据是随机,本应该是小于90个的,但是为了省时间,我就随机造了数据。
紧接着画和弦图:
chordDiagram(mat) ##和弦图命令
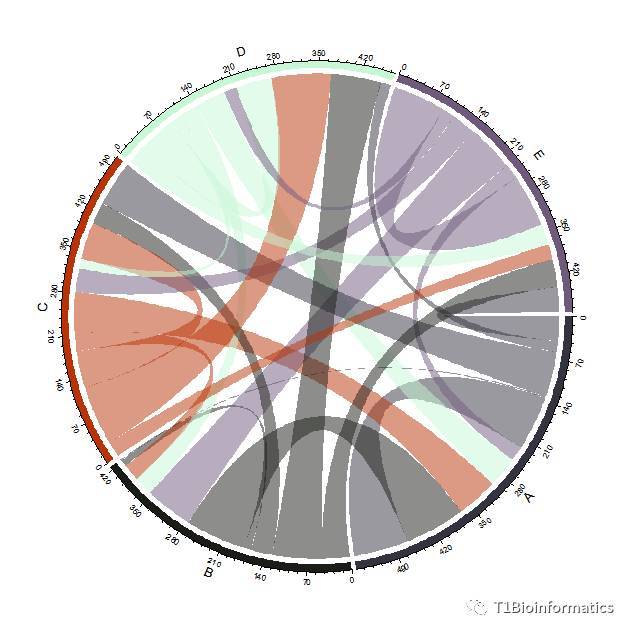
后面我再补充吧,东西太糙了,够用,如果想知道如何添加其他的需求,可以发消息给我。
转载自EasyCharts团队!
- 本文固定链接: https://maimengkong.com/image/1031.html
- 转载请注明: : 萌小白 2022年6月24日 于 卖萌控的博客 发表
- 百度已收录
