ComplexHeatmap是 Zuguang Gu博士开发的一款可以绘制复杂热图的一个包,即可实现简单热图的功能,更能绘制更复杂的热图。复杂的热图有效地可视化不同数据集源之间的关联并揭示潜在模式。
ComplexHeatmap包提供了一种高度灵活的方式来排列多个热图并支持各种注释图形。技术小编之前对这个包做过简单介绍( 如何让你的图变得高大上之COMPLEXHEATMAP ),这次介绍两个简单实用的技巧。
构建测试数据,包括表达量数据、样本分类数据、基因注释分类数据:


技巧1:先分组再聚类
以样本(表达量矩阵中的列)为例,常规操作中可以选择聚类or 不聚类两种,代码如下:
col_fun<-colorRampPalette(c( "navy", "white", "firebrick3"))( 100) #构建用于绘图的颜色
exp<- exp[,match(sample_pdata[, 1],colnames( exp))] #表达量按照样本顺序排个序
col_anno <- HeatmapAnnotation( #构建列的注释信息
group = sample_pdata$group,
col = list(group = c( "B"= "#E31A1C", "CK"= "#007947"))
)
Heatmap( exp,name = "expression",col = col_fun,
column_names_rot = 45, #列名倾斜角度
column_names_centered =TRUE, #列名展示位置
top_annotation = col_anno, #列注释位置top/bottom
cluster_columns = FALSE #列是否聚类,默认TRUE
)
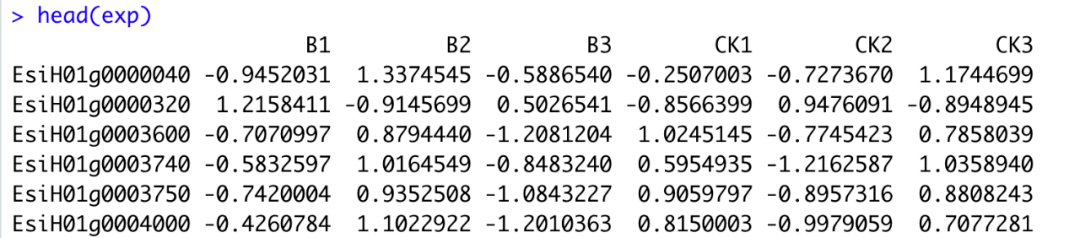
图1A 进行聚类时B2和CK2样本与同组重复样本并不能很好的聚集到一起,图1B不聚类的情况下不能直接看出组内样本的聚集情况(这里为了效果展示,所以选取的数据比较极端,并且先进行了scale 所以会有负值)。
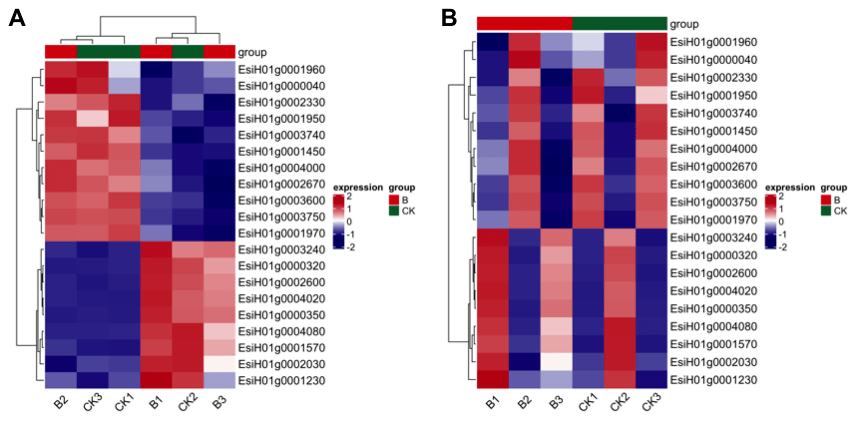
图1
小编只能掏出实用的热图神包ComplexHeatmap了 ~代码如下:
Heatmap(exp,name = "expression",col = col_fun,
column_names_rot = 45,column_names_centered = TRUE,
top_annotation = col_anno,cluster_columns = TRUE,
column_split = sample_pdata$group #分组
你品,你细品,你再 品~ 小编就只多加了最后一行代码 ,图2A可以看出来样本先根据组别分了组,然后组内再进行了聚类。
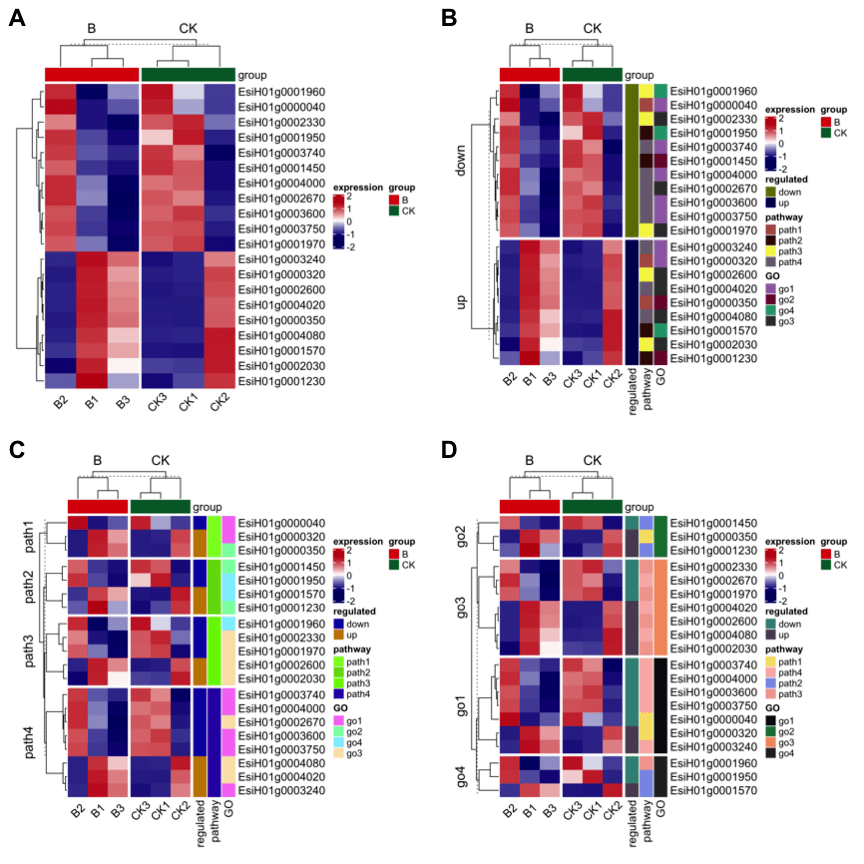
图2
基因同样可以如此操作按照上下调、pathway注释、 Go注释等分组 图2BCD,小编心里又一次给大佬双击666。按照上下调分组的代码如下:
gene_pdata<-gene_pdata[order(gene_pdata$regulated),]
exp<- exp[match(gene_pdata[, 1],rownames( exp)),] #根据基因顺序重新排序
row_anno <- rowAnnotation(
regulated = gene_pdata$regulated,
pathway = gene_pdata$pathway,
GO = gene_pdata$GO
)
Heatmap( exp,name = "expression",col = col_fun,
column_names_rot = 45,column_names_centered =TRUE,
top_annotation = col_anno,right_annotation = row_anno,
column_split = sample_pdata$group,row_split = gene_pdata$regulated
)
技巧2:标签注释
很多情况下,我们绘制热图的基因数目比较多,不便于显示基因名,但是又想突出展示感兴趣的几个基因,ComplexHeatmap 包也考虑到了这个问题,撒花~根据绘图数据的基因原始位置信息进行标签,随便选exp 第一行和第二行的两个基因,构建的行注释文件中添加anno_mark,图3A可以看出来只标记了我们选择的两个基因,代码如下:
row_anno < -rowAnnotation(
regulated= gene_pdata$regulated,
pathway= gene_pdata$pathway,
GO= gene_pdata$GO,
foo= anno_mark(at= c(1,2),labels= c(' EsiH01g0000040',' EsiH01g0000320'))
)
Heatmap( exp, name= "expression", col= col_fun,
column_names_rot= 45,column_names_centered= TRUE,
top_annotation= col_anno,right_annotation= row_anno,
column_split= sample_pdata$group,
show_row_names= FALSE,#是否显示行名,默认TRUE
row_split= gene_pdata$GO
)
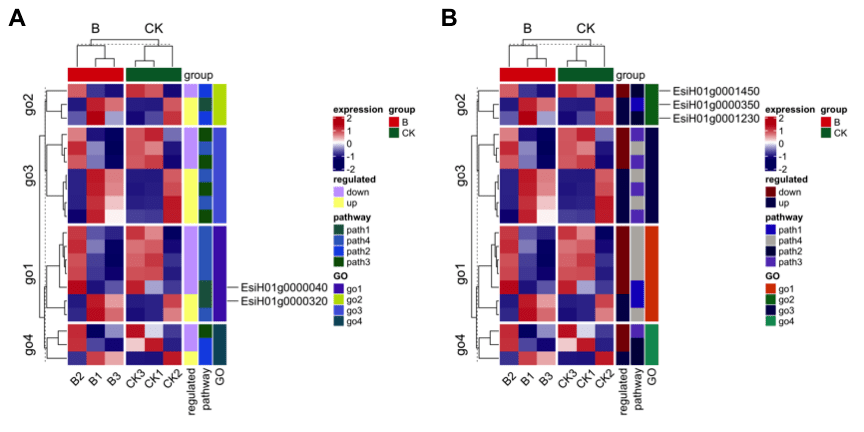
图3
也可以根据基因的分组进行选择展示,比如说提取go2这个功能对应的三个基因位置信息见图3B,代码如下:
hm < -Heatmap( exp, name= "expression",
col= col_fun,
column_names_rot= 45,column_names_centered= TRUE,
top_annotation= col_anno,right_annotation= row_anno,
column_split= sample_pdata$group,
row_split= gene_pdata$GO
)
hm_order<-row_order(hm) #提取位置信息

row_anno < -rowAnnotation(
regulated= gene_pdata$regulated,
pathway= gene_pdata$pathway,
GO= gene_pdata$GO,
foo= anno_mark(
at= c(hm_order$go2),
labels= rownames(exp)[hm_order$go2]
)
)
ComplexHeatmap包还有更多炫酷并且实用的功能,期待大家的进一步发现。
文:CY
排版:市场部
- 本文固定链接: https://maimengkong.com/image/1030.html
- 转载请注明: : 萌小白 2022年6月23日 于 卖萌控的博客 发表
- 百度已收录
