跟着Nature Communication学绘图
之Figure 1的绘制
大家好,我是阿琛。在前面讲解的内容中,我们学习了数据的预处理,构建Seurat对象,以及后续的降维分析处理及其可视化过程。接下来,我们来看下如何对PCA分析得到的结果进行可视化展示,以及GSEA分析。
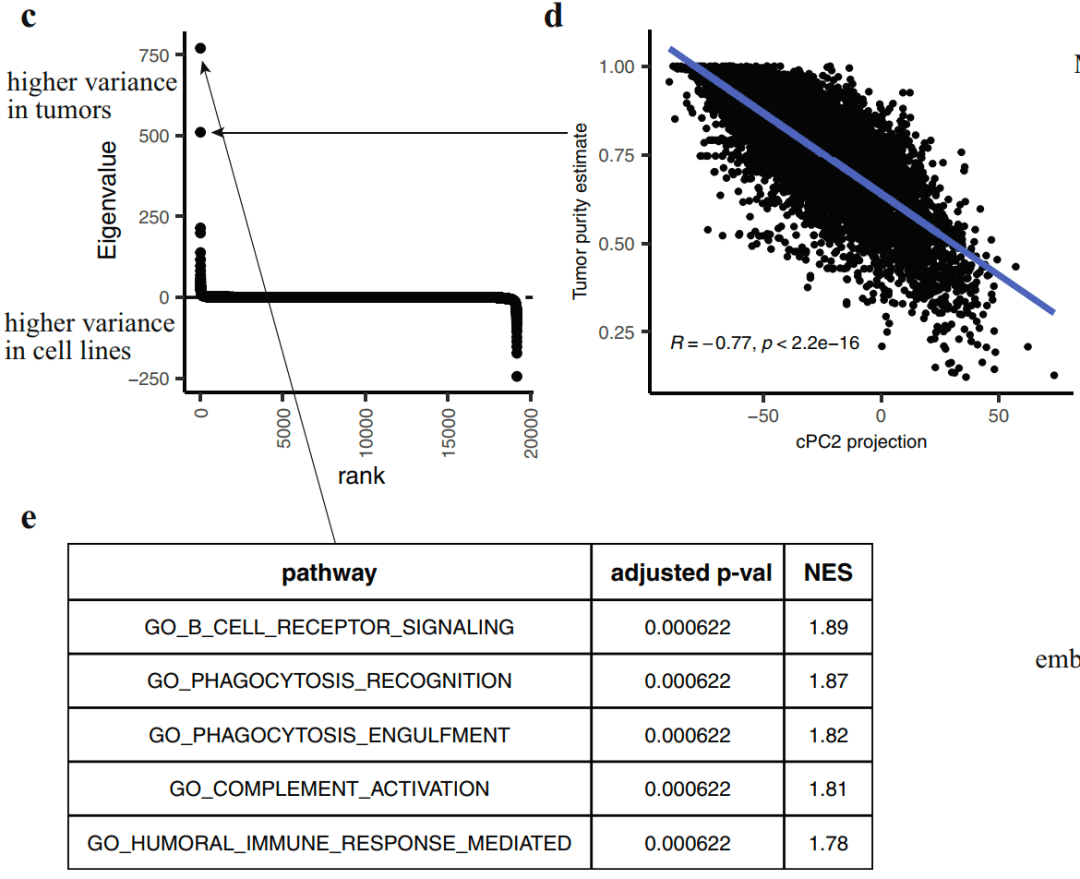
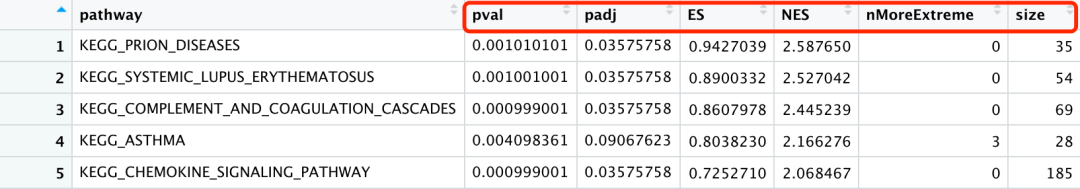
可以看到,在图中,图c为PCA分析得到的cPC 特征值按等级排序,包括19,188 个特征值。图d为肿瘤样本在 cPC2 上的投影与其估计得到的肿瘤组织纯度之间的 Pearson 相关性分析。而在图e中,则为GSEA分析得到的前5个结果。
下面,我们来看一下这几张图的绘制过程。
一、R包加载
library(magrittr) library(tidyverse) library(ggplot2) library(ggpubr) library(fgsea) library(msigdbr) source( "global_params.R") source( "analysis_helpers.R") source( "Celligner_helpers.R")
分析之前,我们需要加载了所需要的R包;由于涉及GSEA分析,因此加载fgsea包。同时,加载相应的自定义函数内容。
二、读取数据
接下来,我们将需要用到的数据读取进来。
cPCA_values<- read.csv( "cPCA_values.csv", header = T)
首先,是PCA降维分析得到的结果值,以用于Figure 1C图的绘制。
alignment <- read.csv( "Celligner_info.csv", header = T) rownames(alignment) <- alignment[,1] alignment[1:3, 1:3]
接着,是相关注释数据的读取。

gene_stats<- read.table( "gene_expression_of_avg&SD.txt", sep = "\t", header = T) gene_stats <- gene_stats[!duplicated(gene_stats $Ensembl),] rownames(gene_stats) <- gene_stats $Ensembl gene_stats[ 1: 3,]
其次,是上期内容分析得到的基因信息文件,包括了基因名称,及其在肿瘤组织和细胞系中表达的平均值和方差值。

load( "result_NC.Rdata")
最后,则是前面处理完成的表达和注释数据。
三、Figure 1C复现
首先,我们来看下如何根据PCA降维结果进行Figure 1C的复现过程。
cPCA_eigenvalues_plot <- data.frame( eval=cPCA_values$cPCA_values, rank = se q(length(cPCA_values$cPCA_values)))
根据cPCA值,同时对其排序进行编号rank,形成新的数据框,以作为绘图的输入数据。
结果显示:
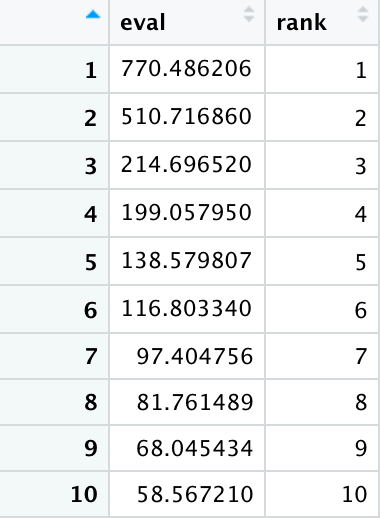
##绘制结果 ggplot(data = cPCA_eigenvalues_plot, aes(x= rank, y = eval)) + geom_point(size=1) + geom_hline(yintercept = 0, linetype = 'dashed') + ylab( 'Eigenvalue') + theme_classic + theme(axis.text = element_text(size=6), axis.title.x = element_text(size=6), axis.title.y = element_text(size=6), axis.text.x = element_text(angle = 90, hjust = 1))
接着,将前面制作完成的cPCA_eigenvalues_plot作为输入数据,使用ggplot函数,进行绘制。
结果显示:
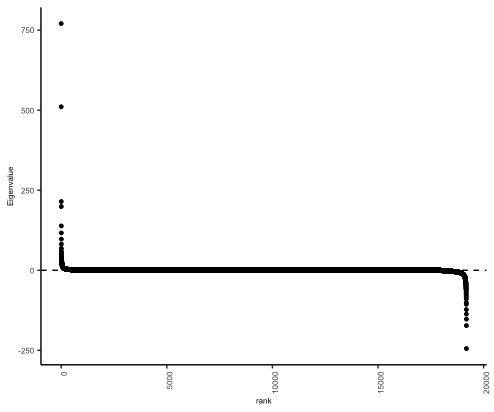
这样,Figure 1C就复现完成了。
四、Figure 1D和Figure 1E输入数据准备
接下来,我们进一步讲解一下Figure 1D和Figure 1E图的内容。但是,在正式的绘图之前,首先需要准备相应的输入数据。
cur_vecs < -read.csv(" cPCs.csv", header= T,row.names= 1) cur_vecs[ 1:3,]
首先,将作者准备好的PCA分析得到的PCs数据读取进来。当然,在自己的分析过程中,大家也可以参考前面几期的内容,通过构建Seurat对象,数据中心化处理,以及PCA降维以得到这部分输入数据。

comgene< -intersect( rownames( cur_vecs), rownames( gene_stats)) cur_vecs< -as.matrix( cur_vecs[comgene,]) gene_stats< -gene_stats[comgene,]
接着,选取输入数据cur_vecs和基因文件gene_stats的共同基因信息,一共得到18164个共同基因。
结果显示:
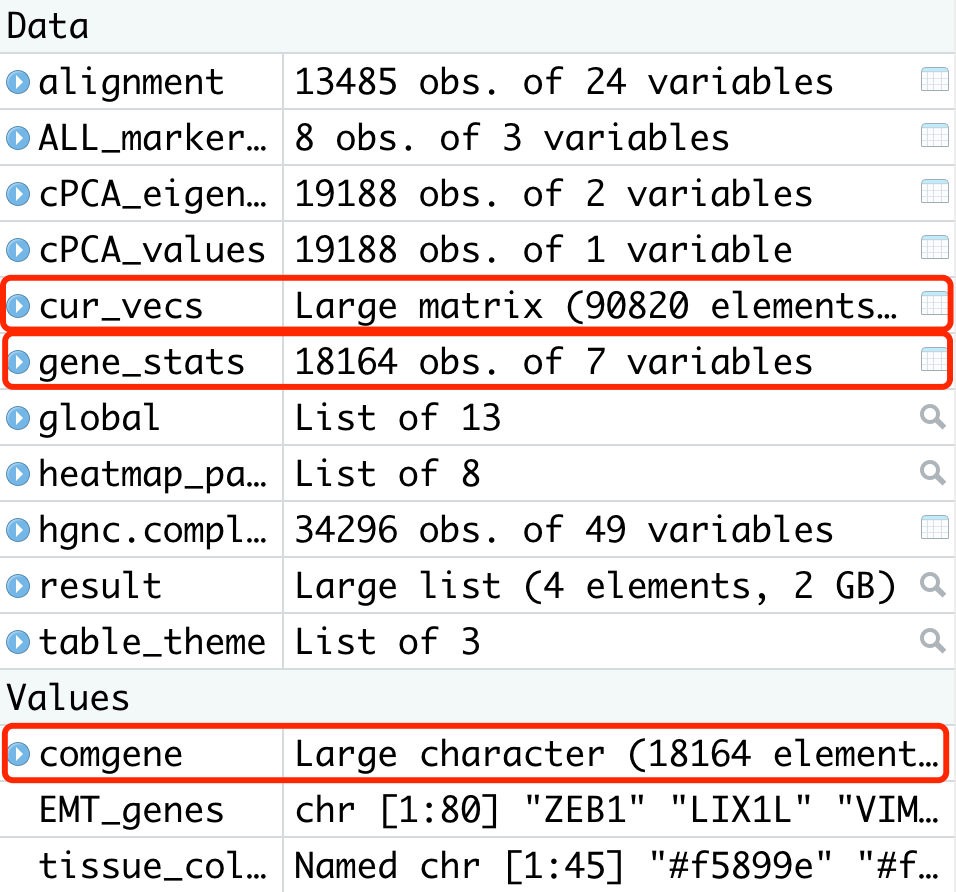
rownames(cur_vecs) <- gene_stats $Gene cur_vecs[1:3,]
随后,将基因名(gene symbol)赋值给cur_vecs的行名。
结果显示:

TCGA_mat<- result $TCGA_mat[,rownames(cur_vecs)] TCGA_ann <- result $TCGA_ann CCLE_mat <- result $CCLE_mat[,rownames(cur_vecs)] tcga_ccle <- rbind(TCGA_mat, CCLE_mat)
提取肿瘤组织和细胞系的表达数据,并进行合并,形成最终的表达数据集tcga_ccle。
结果显示:
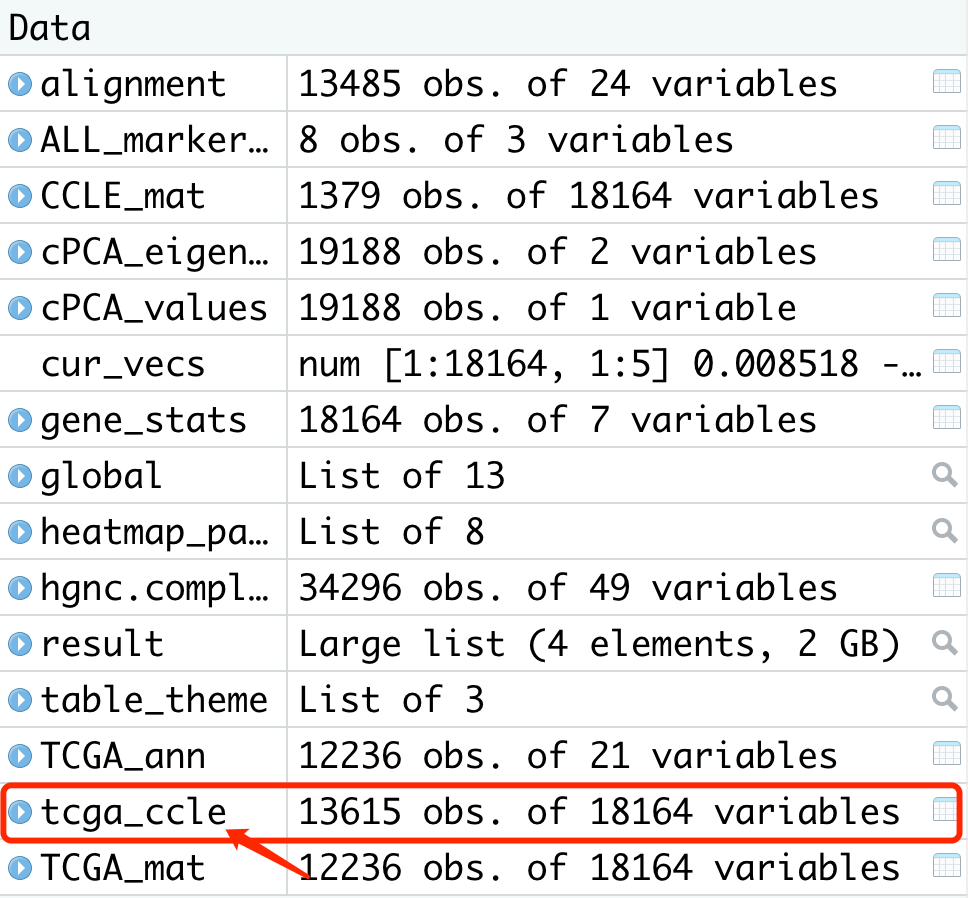
cur_eig_proj<- get_signature_proj(cur_vecs, tcga_ccle, alignment[rownames(tcga_ccle),]) flip_sign<- choose_eig_sign(cur_eig_proj) cur_vecs<- cur_vecs * flip_sign
结果显示:

五、Figure 1D的绘制
dproj<- lm(t(TCGA_mat) ~ cur_vecs) $coefficients[2,] df <- TCGA_ann %>% dplyr::inner_join(data.frame(proj = dproj, sampleID = rownames(TCGA_mat)), by = 'sampleID')
通过lm函数构建线性模型,并提取对于的系数;随后,将得到的结果添加到注释文件TCGA_ann中,以作为Figure 1D绘制的输入数据。
ggplot(df %>% dplyr::filter(!is.na(purity)), aes(proj, purity)) + geom_point(size= 0.5) + ylab( 'Tumor purity estimate') + xlab( 'Eigenvector projection') + stat_cor(label.y.npc = 'bottom', size= 2) + geom_smooth(method = 'lm') + theme_classic + theme(axis.text = element_text(size= 6), axis.title.x = element_text(size= 6), axis.title.y = element_text(size= 6)) + xlab( "cPC2 projection")
结果显示:
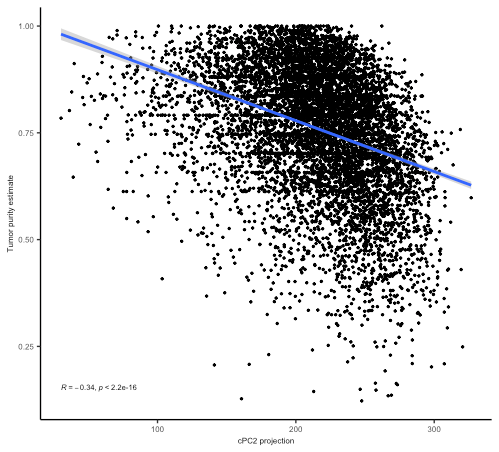
到此,Figure 1D就绘制完成了。
六、Figure 1E数据的准备
最后,我们来看下Figure 1E的内容,涉及了GSEA分析。
df < -data.frame( vec= cur_vecs,Gene= rownames(cur_vecs))%> % dplyr::left_join(gene_stats, by = "Gene") gene_stat < -with( df%> % filter(!is.na(Gene)), gene_stats$Gene)
首先,还是输入数据的准备。将PCA分析得到的PCs与基因信息进行合并,并提取其中的基因名。
mdb_c2 <- msigdbr(species = "Homo sapiens", category = "C2") mdb_kegg = mdb_c2 [ grep( "^KEGG",mdb_c2 $gs_name),] fgsea_sets<- mdb_kegg %>% split( x= .$gene_symbol, f = .$gs_name)
随后,直接使用msigdbr函数下载人类C2注释数据。当然,关于这部分数据,大家也可以直接进入官网(www.gsea-msigdb.org/gsea/msigdb)进行下载。另外,在文章中,作者使用的是GO相关数据信息,大家也可以参考这部分内容进行相应数据的选择。
结果显示:
gene_stats$logFC <- gene_stats$Tumor_mean / gene_stats$CCLE_mean geneList = gene_stats[, 8] names(geneList) <- as.character(gene_stats$Gene) geneList <- sort(geneList, decreasing = TRUE) fgseaRes <- fgsea(fgsea_sets, stats = geneList, nperm = 1000) %>% dplyr::arrange(dplyr::desc(NES)) %>% dplyr::select(-leadingEdge) fgseaRes[ 1: 3,]
最后,使用fgsea包的fgsea函数进行GSEA分析,并提取其中的相对富集分数NES值。
结果显示:

七、Figure 1E表格的绘制
最后,我们对GSEA分析结果进行表格的制作。
cPCA_GSEA_data <- rbind.data.frame(fgseaRes %>% dplyr::arrange(dplyr::desc(NES)) %>% head( 5)) cPCA_GSEA_data$pathway <- factor(cPCA_GSEA_data$pathway, levels = cPCA_GSEA_data$pathway) cPCA_GSEA_data$pathway <- sub( "^([^_]*_[^_]*_[^_]*_[^_]*_[^_]*).*", "\\1", cPCA_GSEA_data$pathway) cPCA_GSEA_data$pathway <- factor(cPCA_GSEA_data$pathway, levels = unique(cPCA_GSEA_data$pathway)) cPCA_GSEA_data <- as.data.frame(cPCA_GSEA_data)
通过选取前5个富集结果,并进行相应的数据清洗。
结果显示:
cPCA_GSEA_data$`adjusted pval` <- signif(cPCA_GSEA_data $padj, 3) cPCA_GSEA_data$NES <- signif(cPCA_GSEA_data $NES, 3)
同时,对adjusted pval值和NES值进行整理,保留3位有效数字。
结果显示:
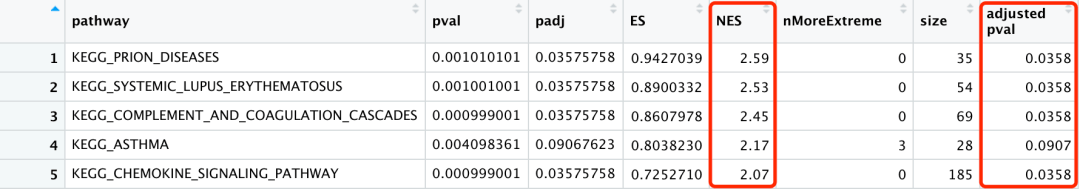
library(gridExtra) grid.table(cPCA_GSEA_data[,c( 'pathway', 'adjusted pval', 'NES')], theme=table_theme, rows= NULL)
使用gridExtra包的grid.table函数对进行表格绘制。
结果显示:

到此,Figure 1的整体内容进行绘制完成啦~赶紧动起手来,大家可以根据这几期的内容进行自己内容的分析。
