Hi,大家好,我是晨曦
根据各位小伙伴的私聊以及前几期推文的阅读量来看,可能Python并不能并不是十分契合现阶段小伙伴的需求,而且也有小伙伴私信晨曦,希望晨曦可以再更新一些单细胞相关的分析内容,说自己常规的细胞轨迹分析、细胞通讯分析以及转录因子分析已经不能满足自己的需求了~
那么好,晨曦接下来会更新一些单细胞新出的高阶分析,争取做一个单细胞高阶分析“百科全书”,也希望各位小伙伴如果觉得内容不错,也欢迎点赞转发哦~
什么是RNA速率(RNA velocity)
复杂的概念并不是我们追求的内容,我们这里简单概括一下:单细胞scvelo分析是一种基于单细胞RNA测序数据的分析方法,用于研究单个细胞之间的转录调控关系和细胞发育轨迹。scvelo是single-cell velocity(单细胞速率)的缩写,它能够探测细胞转录状态的动态变化,预测细胞的未来发育方向和状态,并揭示细胞发育过程中的分子机制。
那么,我们可以明确,这项分析我们可以应用在具有潜在发育组织的样本中
然后我们接下来,来看看别人是如何使用这项分析的,下面笔者将通过文献的方式给各位小伙伴进行高度总结:
1.RNA丰度是单个细胞状态的一个强有力的指标。单细胞RNA测序可以定量、准确、灵敏、准确地揭示RNA丰度。然而,这种方法只能在某个时间点捕获静态快照,这对分析诸如胚胎发育或组织再生等随时间变化的现象提出了挑战。在这里,本文章证明了RNA速率——基因表达状态的时间变率——可以通过区分普通单细胞RNA测序中未剪接和剪接的mRNA来直接进行估计。 RNA速率是一种高维的载体,可以在数小时的时间尺度上预测单个细胞的未来状态。本文在神经嵴谱系中验证了它的准确性,展示了它在多个已发表的数据集和技术平台上的应用,揭示了发育中的小鼠海马的分支谱系树,并检测了人类胚胎大脑中的转录动力学。 RNA速度将极大地帮助分析发育谱系和细胞动力学,特别是在人类。
2.简单理解,我们 常规使用的基因表达矩阵可以理解为是在转录组细胞表达的“瞬间”,但是正常来说, 机体内无时无刻不在进行转录和翻译,也就是转录、剪接和降解,它们都是有速度的,作者在文中就 定义了这些速度,所谓的RNA速率其实就是成熟的mRNA的丰度变化速度,可以通过前体mRNA和成熟mRNA的指标估计,并且可以用来预测细胞未来数小时内的状态
然后笔者在pubmed上检索了一下使用RNA velocity的文献,然后发现了下面这篇文献:
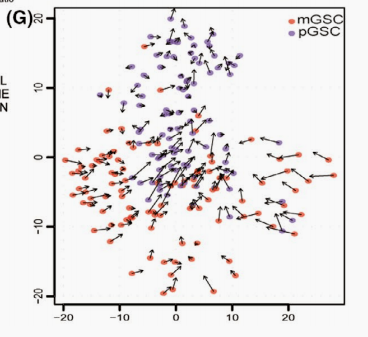
然后这篇文章运用RNA velocity阐释了同一个样本中感兴趣的细胞亚群的分化情况,首先作者提取了单细胞亚群中 mesenchymal GSC(间充质胶质瘤干细胞)和proneural GSC(神经表型间充质干细胞),然后进行了RNA velocity分析揭示了RNA velocity showed that GSC subtype transitions from mesenchymal to proneural phenotype
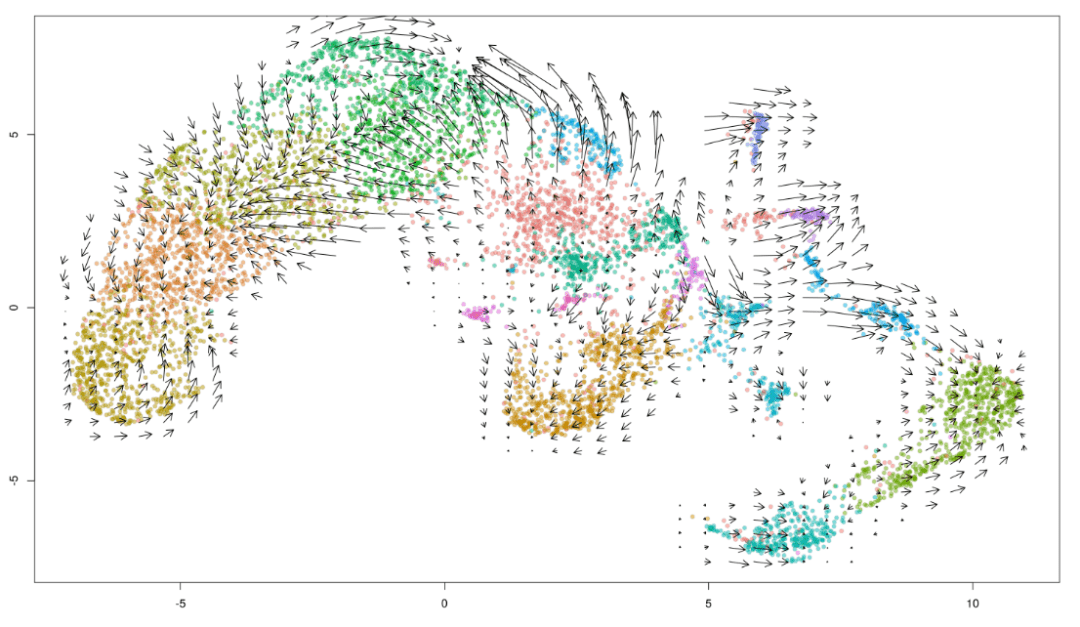
轨迹分析目前来说有两类:
1.拟时序分析
2.RNA velocity
简单来说就是RNA velocity分析并不需要在分析中指定起点和终点,但是输入文件需要我们认为整理成loom文件,这个在后面的代码演示中会着重强调
2.代码演示
然后我们通过上述标准流程的分析即可完成RNA velocity分析,这个分析的代码其实并不是很复杂,难点其实感觉是在需要一定的linux以及python的基础去整理并获得loom文件,好在网上也有质量高的逐步教程,如下:
RNA velocity分析练习(一)文件下载以及预处理 - 简书 (jianshu.com)
然后我们之所以要从上游开始进行是因为我们在进行RNA速率分析的时候,需要区分表达矩阵内前提mRNA(unspliced)和成熟的mRNA(spliced),所以需要从原始数据入手,得到loom文件后,再去进行后续分析自然一切就水到渠成了。
在推文的最后不禁发问:如果是你,你会选择拟时许分析还是RNA velocity分析呢QAQ
我是晨曦,我们下期再见~
参考教程:
1.玩转单细胞高级分析 | 细胞轨迹分析篇 - 知乎 (zhihu.com)
2.单细胞转录组数据分析|| scVelo 教程:RNA速率分析工具 - 简书 (jianshu.com)
3.基于Seruat进行RNA velocity分析:Estimating RNA Velocity using Seurat (htmlpreview.github.io)
4.生成loom文件:RNA velocity分析练习(三)生成loom文件 - 简书 (jianshu.com)
5.比较全面的RNA速率分析单细胞之轨迹分析-6:velocyto.R+Seurat - 简书 (jianshu.com)
6.loom文件的生成:loom文件的生成 - 简书 (jianshu.com)
7.原始代码推文:Estimating RNA Velocity using Seurat (htmlpreview.github.io)
- 本文固定链接: https://maimengkong.com/zu/1532.html
- 转载请注明: : 萌小白 2023年5月13日 于 卖萌控的博客 发表
- 百度已收录
