导读
三代测序和de novo组装技术发展得如火如荼,是如今高通量测序最热门的技术。今天为大家分享一篇三代测序和de novo组装在植物方面应用的综述文章,希望帮助更多的科研小伙伴们了解这项技术,并应用到自己的科研工作中。

发表期刊:Current Opinion in Plant Biology
发表时间:2020.01.22
影响因子:7.508
doi: 10.1016/j.pbi.2019.12.009.
摘要
植物基因组的大小跨越了几个数量级,倍性和杂合度各不相同,并且涉及远古和近代的转座因子扩张,从而使它们的组装非常具有挑战性。单分子测序和光学图谱技术的最新发展,使得复杂度和基因组大小日益增加的的植物物种能够获得高质量的染色体级别的组装本。现在的单分子读长长度可以超过Mb,为解开用二代测序无法读取的基因组区域提供了前所未有的机会。然而,多倍体和高杂合植物基因组仍然难以组装,不过这也为新工具和新方法的产生提供了机会。单倍体分型、结构变异分析和从头组装的泛基因组学是植物基因组组装的新前沿。
引言
在我们庆祝第一个植物基因组(模式植物拟南芥)诞生20周年之际,我们正在进入一个可以将各种大小、复杂程度和倍性的高质量染色体级基因组进行组装的黄金时代。二代测序(NGS)技术(如454和Illumina)以处理更短的序列实现更大的深度,从而使测序难度和测序成本显著降低而引发了植物基因组学革命。如今,单分子测序再次改变了植物基因组组装的格局,首次实现了近乎完整的染色体。
第一个完全基于PacBio单分子实时(SMRT)测序的植物基因组是最小的、也是非常耐干旱的复活草(Oropetium thomaeum),这是包括30%的完整着丝粒在内的当时第四个组装最连续的基因组。三代组装工具Falcon和CANU的开发对PacBio SMRT测序数据的基因组组装具有至关重要的作用。Oxford Nanopore Technologies (ONT)发布的第一个纳米孔测序仪,其读长超过了PacBio,其中一些酶的读长超过了Mb,并且能够装配更多的拟南芥、番茄、高粱、香蕉和十字花科植物的相邻和完整的参考基因组。在过去20年里已经发表的植物基因组近400个,包括333个被子植物、15 个裸子植物、 2个轮藻和44个绿藻。
本综述的关注点在于过去几年中,长reads测序和能够组装近乎完整基因组的技术进展。
长读长单分子测序
长reads测序技术的发展是推动植物基因组组装的主要动力 (图1)。由于植物基因组中存在高杂合度、复杂多倍体和高度重复序列,对植物基因组的组装被证明是最具挑战性的。一般读长必须超过在基因组中发现的显性重复长度,并且可以跨越20 - 200kb的巢式长末端重复(nested LTR)或单倍体区段。
不同厂商的长reads读取技术各有千秋。如PacBio是第一个提供长reads读取的系统,一般N50大于20 kb。ONT随后发布的MinION和PromethION测序仪,其N50s在20~50 kb,有些长度超过了几Mb。长reads的优点是可以顺利跨过基因组上高复杂或高重复区域,获得更完整的组装效果。但其缺点也不容忽视:(1)需要抽提高质量的DNA,而且reads 越长,对DNA的质量和完整度要求越高。(2)更高的错误率。对此,PacBio通过更新CCS的方法来解决错误率问题,采用几近完美的HiFi 15 kb的读长模式,其准确度高达99.8%。不过高质量也导致相应成本的增加,而且,即使是几近完美的15 kb读长也可能无法实现复杂植物基因组中常见的巢式的、高度相似的重复结构。许多复杂的植物基因组具有大于20kb的重复结构。
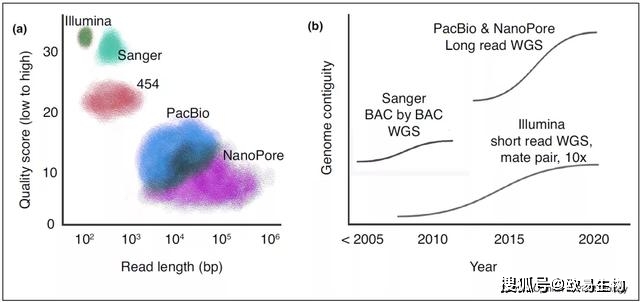
图1 | 过去二十年长读长测序技术的发展
长读长易错基因组组装
根据计算机的设计、速度和内存使用,以及物种的复杂性、杂合性、多倍体或大基因组等因素,所用的算法也各不相同。大多数主要的组装程序,如CANU、Falcon (phase和unzip)、MARVEL和MECAT,都采用了一种自我校正的方法,在这种方法中,长reads相互比对,并用足够高的覆盖率来校正错误。相比之下,一些长reads组装程序是“无校正”的,比如OLC-based minimap2/miniasm,或者可以利用校正reads,比如基于BDG-based WTDBG2和Flye。对于一些高度复杂的基因组,如大麻属植物,四氢大麻酚合成酶基因(THC)嵌套在70 - 90kb的LTR-based串联重复序列中,需要无校正组装。草图长reads组件有残留错误,必须使用高覆盖率长reads和/或短reads数据的组合进行polish。Quiver/Arrow (PacBio)、Medaka (ONT)、Nanopolish和Racon被设计用于长reads数据,Pilon使用Illumina数据进行polish。在一般情况下,建议对长reads组装程序进行三轮或更多的consensus和polish,以达到>99.6%的精度。但最近PacBio CCS HiFi reads的读取,利用一种基于Flacon的Peregrine新组装程序,时间和内存消耗都是Falcon组装subreads的一小部分,极大地提升了组装效率。
辅助组装技术
单分子reads产生的草图具有很高的连续性,但染色体级的组装是标记辅助育种、定量遗传学和比较基因组学所需要的。高密度的遗传图谱传统上被用来将contigs和scaffolds固定在染色体中,但它们容易出现排序和定位问题。遗传图谱也不能锚定重组率低或标记密度低的序列,并且需要构建分离群体。目前主要的scaffolding方法依赖于染色质的远程互作或光学图谱进行跨染色质锚定(图2)。Hi-C依赖于从交联染色质到定向互作的密度和邻近性。染色体占据细胞核的不同区域,染色体内相互作用比染色体间相互作用更容易发生。染色质相互作用的概率随线性距离衰减,而长程相互作用(>100 Mb)是罕见的,但比染色体间接触更频繁。这一原理可用于可靠地创建一个Hi-C相互作用矩阵,用于相邻区域的排序和scaffold即使是小的或重复的contigs。尽管通常情况下Hi-C能够将基因组组装到染色体级别,甚至对于异源四倍体如Eragrostis tef 也能实现,但对更复杂的植物基因组仍可能存在问题。另一项有望实现的物理测绘技术是光学图谱,它可以通过纳米通道拉伸标记的DNA分子。光学绘图方法在技术上具有挑战性,需要对每个物种进行优化。
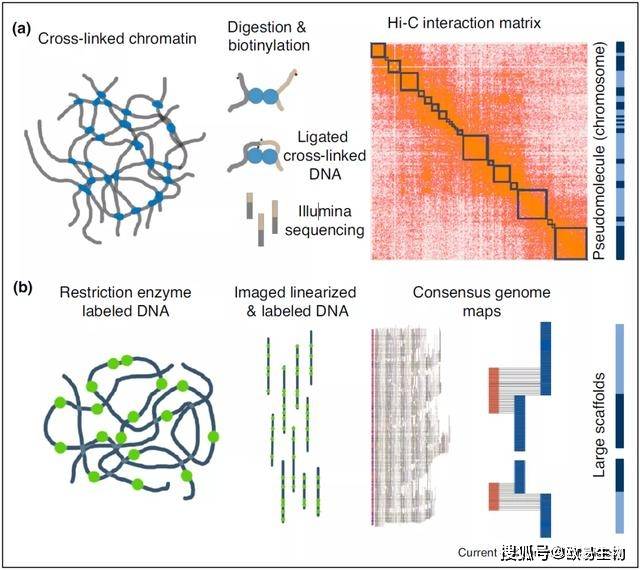
图2 | 长reads拼接组装的主要策略
复杂植物基因组解析
单倍体或单倍体参考基因组简化了下游分析,但未能获得个体的真实基因组组成。玉米和拟南芥等近亲繁殖的物种是高度纯合,但许多异交物种和无性繁殖的高度杂合物种,同源染色体之间具有大量的重复元件、SNP和的结构变异(SV)。单倍型的精确组装和定相(phasing)对于等位基因特异性分析杂种优势和亚基因组显性等复杂性状以及具有生物学或农学重要性的杂合位点的克隆至关重要。长reads组装算法可以准确地纠正和解决不同的单倍型,使组装体超过单倍体基因组的大小(图3)。这些基因组有一个混合的“重复”区域,其中不同的单倍型分别组装,将单倍型与少数多态性各种的单拷贝区域整理成一个。在对基因剂量和重复开展下游分析,以及表达定位、重测序、甲基化或其他数据分析之前,必须标记重复区域。部分分型基因组可以塌缩成一个嵌合单倍体,分型成完整的单倍体,或者用一个基于图形的组装结构将单倍体维系为一个混合物(图3)。FALCON-Unzip被用来创建一些葡萄物种的分型二倍体的组装;将10× Genomics与长reads和高覆盖度的Illumina二代测序数据相结合,组装和分型了同源四倍体蓝莓的所有四个单倍体[53]。PacBio长读长和Hi-C的应用使得八倍体甘蔗、复杂异源四倍体花生、埃塞俄比亚画眉草(teff)以及糜子的基因组组装和分型成为可能。
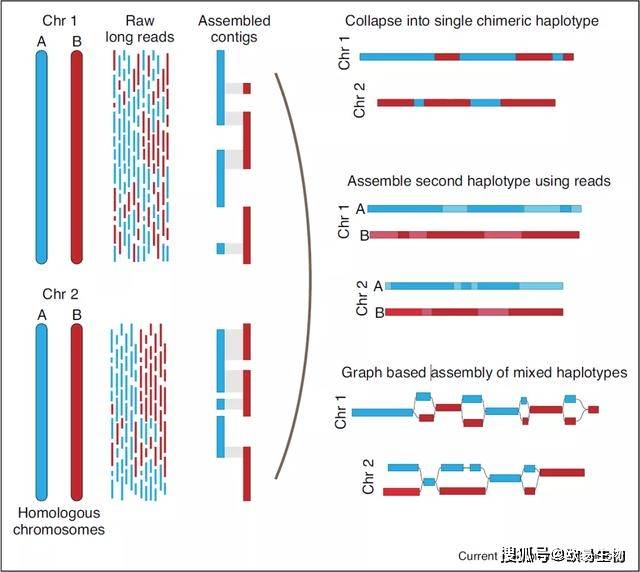
图3 | 杂合基因组测序和分型的组装方法
基因组组装图的利用
基因组草图的组装质量通常由包含>50%的序列长度的contig N50或最短序列长度来评估。“最佳”组装本通常是通过调整参数或测试不同的算法,以产生最高的N50。这种方法存在的问题在于,这种过于简单的度量方法忽略了在组装overlap时遇到的歧义。基因组组装图可以用来可视化相邻contigs之间的复杂性和overlap(图4)。理想的图应该是每个contig(节点)只有一条边,连接表示沿着一条染色体的相邻序列,这通常是拟南芥这样的简单纯合基因组的情况。组装后的单倍体在图上形成“气泡”,其中一个overlap区域连接到两个相邻的分型(phased)单倍体,这通常是林木基因组具有高杂合度和古老重复的情况。复杂序列和高拷贝重复(LTRs、着丝粒等)可以产生成千上万个没有清晰路径的相互连接的节点组成的看不见的“毛发球”。基因组图可以帮助确定在未来的组装迭代中应该修改的参数,或者测试是否需要更多的覆盖率或其他技术来解决组装问题。最终,基因组图可能是一种更好的表示基因组复杂性的方式,尤其是当参考基因组的概念被泛基因组所取代的情况下。
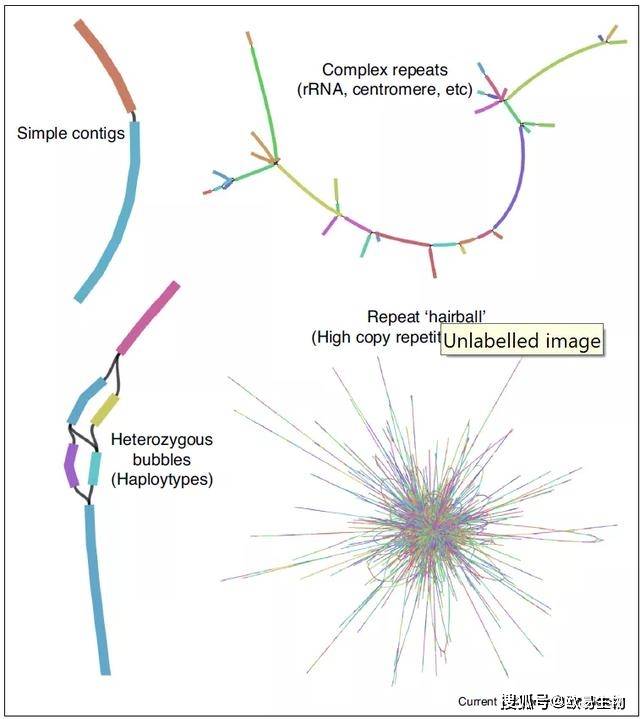
图4 | 用基因组组装图评估基因组质量
当前挑战和未来展望
多倍体和杂合度是基因组组装中不断面临的挑战,但完整、无gap和完全分型的植物基因组即将问世。单分子测序的产能随着成本的下降而上升,和2010年代早期在NGS中观察到的趋势如出一辙。这不仅有助于对几乎所有植物物种进行测序,而且还能在单个物种中产生大量新的参考基因组,以捕捉真正的遗传多样性。
在未来几年内,de novo 组装将取代全基因组重测序应用于人类遗传学和泛基因组分析。基因注释方面的进展一直滞后于基因组组装方面的进步,而产生准确的基因预测仍然是一个主要限制因素。提高注释质量不仅需要纳米孔全长cDNA测序或PacBio Iso-seq等新技术,还需要新算法以更好地预测功能基因组元件。
参考文献
Michael TP, VanBuren R. Building Near-Complete Plant Genomes. Current Opinion in Plant Biology 2020, 54:26–33
END
转自欧易生物
- 本文固定链接: https://maimengkong.com/zu/1995.html
- 转载请注明: : 萌小白 2025年7月27日 于 卖萌控的博客 发表
- 百度已收录
