原文
Zou C, Wang P, Xu Y. Bulked sample analysis in genetics, genomics and crop improvement[J]. Plant biotechnology journal, 2016.
Perspectives

BSA既有前面讨论的一些优势,也存在一些限制。尽管BSA已经广泛应用于遗传学和基因组学研究,但其主要集中在主要基因控制的相对简单的性状上。对于由具有不同效应的基因控制的数量性状,可通过环境型鉴定辅助的精确表型鉴定改进混样的选择,并在环境受控、管理良好的条件下进行实验[1],通过该实验可以对具有相对较大影响的基因进行定位。对于涉及许多基因的复杂性状,每种基因具有较小的作用并且受环境的显著影响,BSA可能不像整个群体的分析一样有效,因为从大量位点选择含有所有有利等位基因的极端表型是不可能的[2]。与遗传分析中的传统方法相比,BSA需要仅识别在目标群体中显示对比极端表型的个体,而不考虑其余性状的准确值[3]。因此,BSA的遗传分析可能抬高或降低实际值,如表型方差,LOD评分和加性效应[4]。此外,BSA一直无法识别上位相互作用[5],并且对偶尔的表型错误更不敏感[6]。通过使用多个混样、精确的表型和环境型鉴定,增加群体大小和标记密度,可以改进BSA的效果[7]。此外,单个基因型的GWAS也可用于验证BSA结果,并能更可靠地阐明复杂场景。
随着基因组测序的成本显着降低,特别是RNA-seq的发展,基因分型比以前更加简化。因此,BSA可以在一个大群体中的几个混池中进行基因型鉴定以提高检测的能力[7,8,9],可以在确定适当的子池之后,在多个批次中或通过个体基因分型来验证感兴趣的罕见事件或等位基因。当涉及NGS辅助的BSA设计优化时,应考虑混池数量、每个池中的个体数及测序深度[10]。与DNA芯片相比,在蛋白质芯片的开发过程中将面对更多的步骤与挑战[11,12,13,14,15,16]。技术挑战是找到一种表面和一种附着方法,使蛋白质保持其二级结构或三级结构,并产生具有长保质期的阵列,使芯片上的蛋白质在短时间内不变性。实验上的挑战是针对靶基因组中的每种蛋白质获得抗体或其他捕获分子,定量结合蛋白的水平,从芯片中提取检测到的蛋白质进行进一步分析,并减少捕获剂的非特异性结合(https://en.wikipedia.org/wiki/Protein_microarray)。最后,应显著增加蛋白芯片的容量,检测整个蛋白质组,并能够检测到低丰度的蛋白质。
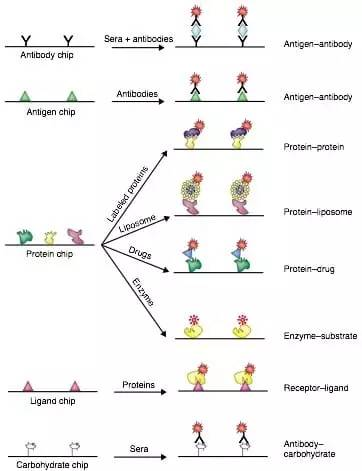
Applications of protein microarrays[28].
从技术层面来讲,基于芯片的高通量蛋白质与蛋白质及其他生物分子多重互作分析具有极大的潜力,也是信号转导途径和网络研究的另一种有力方法[17]。配备多路复用技术,定量蛋白质组学进入“超”高通量时代,使得有可能在一天内全面比较人类所有主要组织/器官[18]或植物。
尽管BSA已被广泛应用于DNA和RNA水平研究,但其蛋白质水平的使用尚未见报道。由于许多基于MS的蛋白质定量工具被设计用于两个样品之间的比较,预计它们中的许多适用于基于蛋白质的BSA。基于蛋白质的BSA的主要挑战是在纤维素和许多植物的次生代谢产物中富集的如多酚,脂质,有机酸,萜烯或色素使得难以从组织中提取纯的蛋白质。
传统的基因分型方法,例如基于单个标记或芯片标记的,应该由GBS来补充,包括常规全基因组测序和简化的基因组测序进行目标富集或降低基因组复杂性[19,20,21,22]。GBS技术灵活高效,为目前可用基因分型技术成本的三分之一[23],提供了可用于基因筛选或GWAS的标记密度,若样本复用则成本可进一步降低[24]。GBS可用于GWAS、基因组多样性研究、遗传连锁分析、分子标记发现和大规模植物育种计划中的基因组选择[25]。当包含在两个混池中的个体数量足够大时,例如超过500个,BSA可以与GBS技术结合使用,并用于GWAS[26,27]。
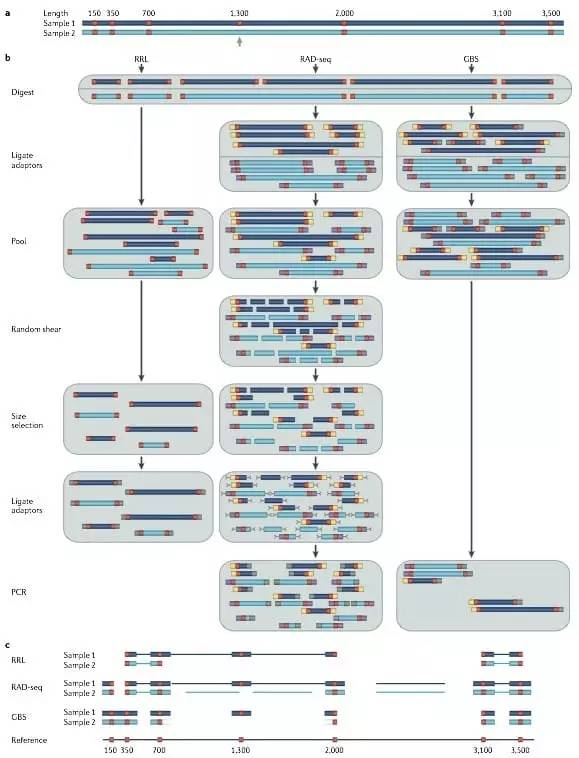
Methods for high-throughput marker discovery[29].
可以预见,BSA在遗传学、基因组学和作物改良中变得越来越重要,并且将在许多情况下取代所有个体(全部群体)的分析。BSA-seq(BSA by sequencing)将随着其数据分析专用软件的开发而变得越加有吸引力,用于分析低频或稀有突变、准确估计单倍型,新的更长读长的测序技术的应用以促进单倍型信息的重建等[27]。由于基因组范围的selective genotyping和BSA成为可能,需要有效的信息管理和数据分析系统帮助其在遗传学、基因组学和植物育种中的应用。
Reference[1] Xu Y. Molecular dissection of complex traits: practice[J]. Molecular plant breeding, 2010: 249-285.
[2] Feng J, Long Y, Shi L, et al. Characterization of metabolite quantitative trait loci and metabolic networks that control glucosinolate concentration in the seeds and leaves of Brassica napus[J]. New phytologist, 2012, 193(1): 96-108.
[3] Yang Z, Huang D, Tang W, et al. Mapping of quantitative trait loci underlying cold tolerance in rice seedlings via high-throughput sequencing of pooled extremes[J]. Plos one, 2013, 8(7): e68433.
[4] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for tranomics[J]. Nature reviews genetics, 2009, 10(1): 57-63.
[5] Schneeberger K, Ossowski S, Lanz C, et al. SHOREmap: simultaneous mapping and mutation identification by deep sequencing[J]. Nature Methods, 2009, 6(8): 550-551.
[6] Soller M, Beckmann J S. Marker-based mapping of quantitative trait loci using replicated progenies[J]. Theoretical and Applied Genetics, 1990, 80(2): 205-208.
[7] Takagi H, Uemura A, Yaegashi H, et al. MutMap‐Gap: whole‐genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii[J]. New Phytologist, 2013, 200(1): 276-283.
[8] Giovannoni J J, Wing R A, Ganal M W, et al. Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations[J]. Nucleic Acids Research, 1991, 19(23): 6553-6568.
[9] Holland J B. Genetic architecture of complex traits in plants[J]. Current opinion in plant biology, 2007, 10(2): 156-161.
[10] Kim S, Kim C W, Park M, et al. Identification of candidate genes associated with fertility restoration of cytoplasmic male-sterility in onion (Allium cepa L.) using a combination of bulked segregant analysis and RNA-seq[J]. Theoretical and Applied Genetics, 2015, 128(11): 2289-2299.
[11] Cronn R, Liston A, Parks M, et al. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology[J]. Nucleic acids research, 2008, 36(19): e122-e122.
[12] Gallais A, Moreau L, Charcosset A. Detection of marker–QTL associations by studying change in marker frequencies with selection[J]. Theoretical and Applied Genetics, 2007, 114(4): 669-681.
[13] Hancock A M, Brachi B, Faure N, et al. Adaptation to climate across the Arabidopsis thaliana genome[J]. Science, 2011, 334(6052): 83-86.
[14] Magwene P M, Willis J H, Kelly J K. The statistics of bulk segregant analysis using next generation sequencing[J]. PLoS Comput Biol, 2011, 7(11): e1002255.
[15] Liu S, Yeh C T, Tang H M, et al. Gene mapping via bulked segregant RNA-Seq (BSR-Seq)[J]. PLoS One, 2012, 7(5): e36406.
[16] Zhu, H. and Snyder, M. (2003) Protein chip technology. Curr. Opin. Chem. Biol. 7, 55–63.
[17] Gallais, A., Moreau, L. and Charcosset, A. (2007) Detection of marker QTL associations by studying change in marker frequencies with selection. Theor. Appl. Genet. 114, 669–681.
[18] Pla-Roca M, Leulmi R F, Tourekhanova S, et al. Antibody colocalization microarray: a scalable technology for multiplex protein analysis in complex samples[J]. Molecular & Cellular Proteomics, 2012, 11(4): M111. 011460.
[19] Henegariu O, Heerema N A, Dlouhy S R, et al. Multiplex PCR: critical parameters and step-by-step protocol[J]. Biotechniques, 1997, 23(3): 504-511.
[20] Popescu S C, Popescu G V, Bachan S, et al. Differential binding of calmodulin-related proteins to their targets revealed through high-density Arabidopsis protein microarrays[J]. Proceedings of the National Academy of Sciences, 2007, 104(11): 4730-4735.
[21] De Godoy L M F, Olsen J V, Cox J, et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast[J]. Nature, 2008, 455(7217): 1251-1254.
[22] El-Soda M, Malosetti M, Zwaan B J, et al. Genotype× environment interaction QTL mapping in plants: lessons from Arabidopsis[J]. Trends in Plant Science, 2014, 19(6): 390-398.
[23] De Godoy L M F, Olsen J V, Cox J, et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast[J]. Nature, 2008, 455(7217): 1251-1254.
[24] Zhang X, Pérez-Rodríguez P, Semagn K, et al. Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs[J]. Heredity, 2015, 114(3): 291-299.
[25] Henegariu O, Heerema N A, Dlouhy S R, et al. Multiplex PCR: critical parameters and step-by-step protocol[J]. Biotechniques, 1997, 23(3): 504-511.
[26] Dupont F M, Vensel W H, Tanaka C K, et al. Deciphering the complexities of the wheat flour proteome using quantitative two-dimensional electrophoresis, three proteases and tandem mass spectrometry[J]. Proteome Science, 2011, 9(1): 10.
[27] Schneeberger K. Using next-generation sequencing to isolate mutant genes from forward genetic screens[J]. Nature Reviews Genetics, 2014, 15(10): 662-676.
[28] Zhu H, Snyder M. Protein chip technology[J]. Current opinion in chemical biology, 2003, 7(1): 55-63.
[29] Davey J W, Hohenlohe P A, Etter P D, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing[J]. Nature Reviews Genetics, 2011, 12(7): 499-510.- 本文固定链接: https://maimengkong.com/zu/1216.html
- 转载请注明: : 萌小白 2022年10月2日 于 卖萌控的博客 发表
- 百度已收录
