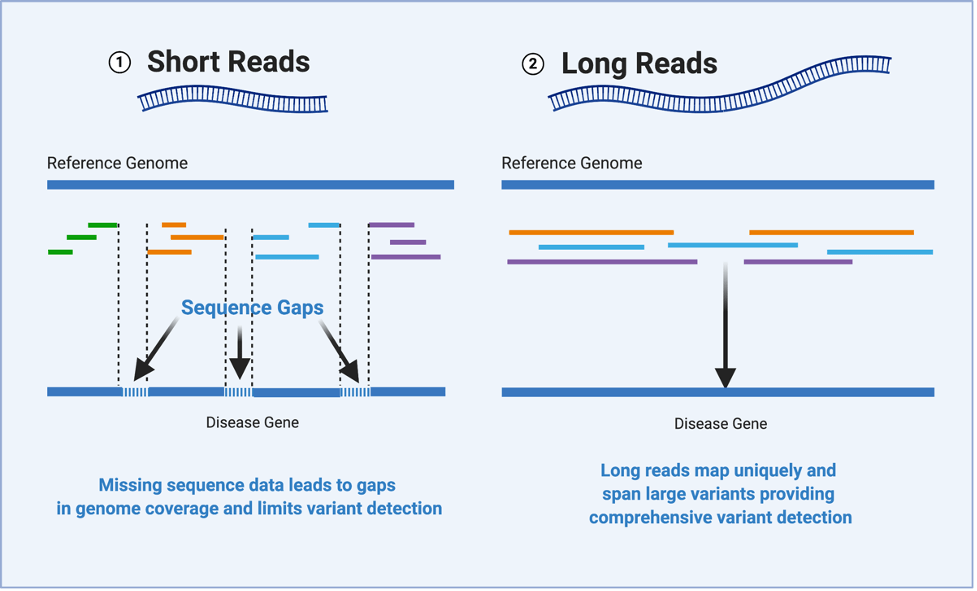
目前,二代测序技术(next-generation sequencing,NGS)凭借其高通量、高准确性与低成本的优势在基因组测序市场占据主导地位。该技术将基因组打断为小片段,在对每个片段进行测序后,生成一小段DNA序列,即读数(reads)。但是,这样的方法会导致基因组数据高度碎片化,产生不完整甚至错误的组装。
近年来,对读长的更高需求加速了长读长测序(long-read sequencing,LRS)技术的发展。LRS技术与短读长测序技术相比,无需将DNA片段化便可进行测序,从而跨越整个重复序列并实现连续和完整的组装。随着测序通量和准确性的提升,LRS技术可以测定上万至几兆碱基的连续序列,技术成熟度稳步提升。目前常使用的是以PacBio SMRT为代表的实时单分子测序技术和以ONT为代表的纳米孔测序技术,它们均能跨越基因组中高重复性的区域,以实现核酸分子更精确完整的检测,识别基因组中大片段的变异和更多的转录本。
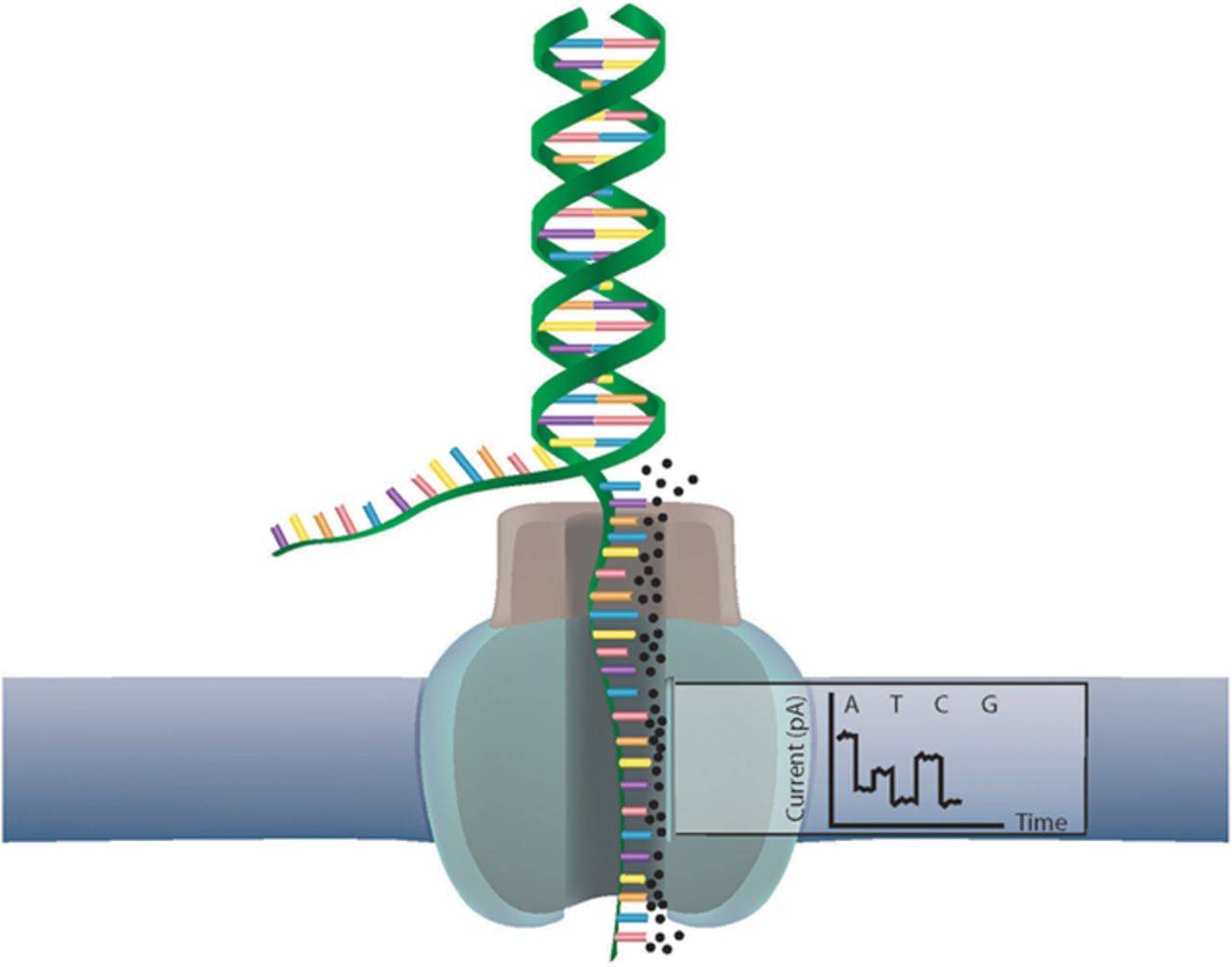
1 LRS技术原理
1.1 PacBio测序技术
PacBio SMRT测序技术采用了一种拓扑环状的DNA分子模板,称为SMRTbell。该模板由两端带有单链发夹式接头的双链DNA插入物组成。组装好的SMRTbell与DNA聚合酶结合,并被装载到包含多达800万个零模波导孔(zero-mode waveguide,ZMW)的SMRT CELL芯片上。在测序反应过程中,聚合酶围绕SMRTbell进行反应,用荧光标记的脱氧核苷三磷酸合成新生链。在每次掺入后,激光将会激发荧光基团,并利用相机记录荧光信号(图1A)。
2019年,PacBio开发了循环共识测序(circular consensus sequencing,CCS),该方法通过从单个模板分子的多次检测中得出共有序列,进而实现高碱基准确度,在长度约为13 kb的序列中碱基准确度超过99%。但该测序过程需要约10 µg DNA样本,对检测样本量有限的微小肿瘤或早期肿瘤造成了一定的困难。同时,这种测序方式虽然准确性比较高,但是并未摆脱对光学系统地依赖;同时由于边合成边测序的基本技术原理,仪器的体积比较大,搬运和携带仍然比较困难。
1.2 纳米孔测序技术
2012年,牛津纳米孔技术公司(Oxford Nanopore Technologies,ONT)公布了首个高通量测序平台MinION。该测序平台先将测序接头与马达蛋白连接于双链DNA分子上,当双链DNA分子进入嵌在合成生物膜上的纳米孔后,马达蛋白即解开DNA双链,带负电荷的DNA在电场力与马达蛋白作用下以可控的速率通过纳米孔。DNA分子在纳米孔中穿过时,会造成电流的扰动,通过记录电流序列依赖性变化,根据电子信号产生的差异识别不同的碱基,从而实现实时分析DNA链中的序列,该技术建库起始量低至2 µg(图1B)。
MinION 和 PromethION 是ONT最常用的两种纳米孔测序仪,MinION 是一种纳米孔型便携式测序仪,最初商业化设备的读取长度约为 10 kb,单个流通池(Flowcell)的吐量约为 5 Gb。PromethION 增加了每个流通池的孔数,并支持运行多个流通池,最多可同时运行 48 个流通池,测序数据量可超过 100 Gb。基于纳米孔测序原理的测序仪特点是体积小巧,具有很好的便携性,适用于条件受限的特殊的应用场景。
近年来,国产纳米孔测序仪也逐步走向市场。齐碳科技有限公司推出了国内首个具有完全自主知识产权的纳米孔测序仪产品QNome-3841,可实现生物芯片、集成电路等核心组件100%国产化。今是科技有限公司推出了国内首款中通量纳米孔基因测序仪Gseq500,单芯片测序单元数量达到50万个。此外,普译生物科技有限公司正在进行具有自主知识产权的纳米孔蛋白优化以及中高通量纳米孔基因测序仪的产品开发。
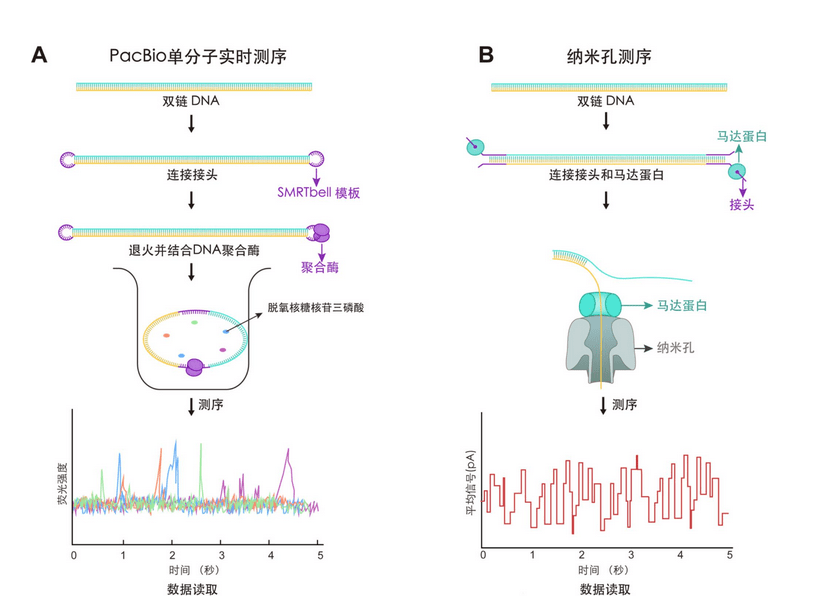 图1 PacBio与ONT测序技术工作原理
图1 PacBio与ONT测序技术工作原理
1.3 新型单分子LRS技术
上述两种测序技术为目前比较成熟的LRS技术,随着研究人员对测序原理的改进,新的单分子LRS技术应运而生,并且能够分析经过修饰的核酸分子。DiMeLo-seq(Directed Methylation with Long-read Sequencing, DiMeLo-seq)结合了抗体靶向的蛋白-DNA检测方法,将靶蛋白结合位点附近的DNA原位甲基化修饰后,使用LRS技术直接读取这些外源性甲基化标记。该方法在检测目标蛋白在单个DNA分子上的多种结合状态上具有独特优势,建库需要3 µgDNA样本,并且可以同时检测外源性抗体靶向的腺嘌呤甲基化和内源性的单分子CpG甲基化,从而实现在高度重复的基因区域内绘制蛋白质-DNA相互作用图谱。
单管长片段测序 (single tube Long Fragment Read, stLFR)是由华大智造科技股份有限公司自主研发的基于DNBSEQ平台的一种长片段读取技术,该方法利用Tn5转座酶将带有分子标签的杂交序列插入DNA中,以对每个长DNA分子进行编码,实现读取序列长度高达10k-300k,建库起始量低至1 ng。
TELL-seq是一种使用NGS技术获取远程区域信息的方法,该方法使用转座酶来片段化DNA并添加条形码,进而帮助NGS技术重新组装DNA片段,让短读长数据产生超LRS效果,等价读长达20 kb-200 kb。与 PacBio 和 ONT 测序方法比,碱基准确度更高,所需的DNA样本量更少,人类基因组DNA仅需5 ng。但需要根据目标基因组进行复杂的生物信息学分析,以对获得的碎片信息进行精确注释。
通过 NGS平台,使用测序技术对转座酶可及性染色质(transposase-accessible chromatin)进行单细胞测定(即scATAC-seq),是一种成熟的对单个细胞内开放染色质区域进行检测的方法。然而,由于基于NGS技术,该方法对检测基因组结构变异存在困难。北京大学汤富酬团队开发了一种基于LRS技术的转座酶可及性染色质单细胞测定方法(scNanoATAC-seq),可以实现同时检测单个细胞内的染色质可及性和结构变异。
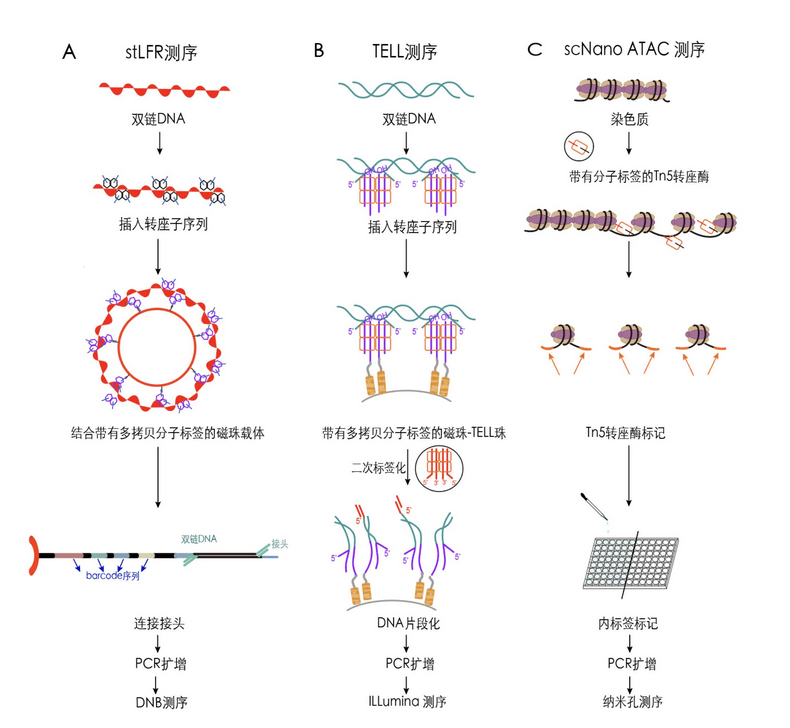 图2 3种新型单分子LRS技术工作原理
图2 3种新型单分子LRS技术工作原理
由于测序方式和DNA样品制备的差异,每一种LRS技术均可以产生不同类型的长序列数据,它们的长度和准确性都不同。在表1中,我们对长读长数据集特征进行总结整理,并与Illumina NGS数据进行比较,重点对比每种测序数据类型的相对长度和精度。
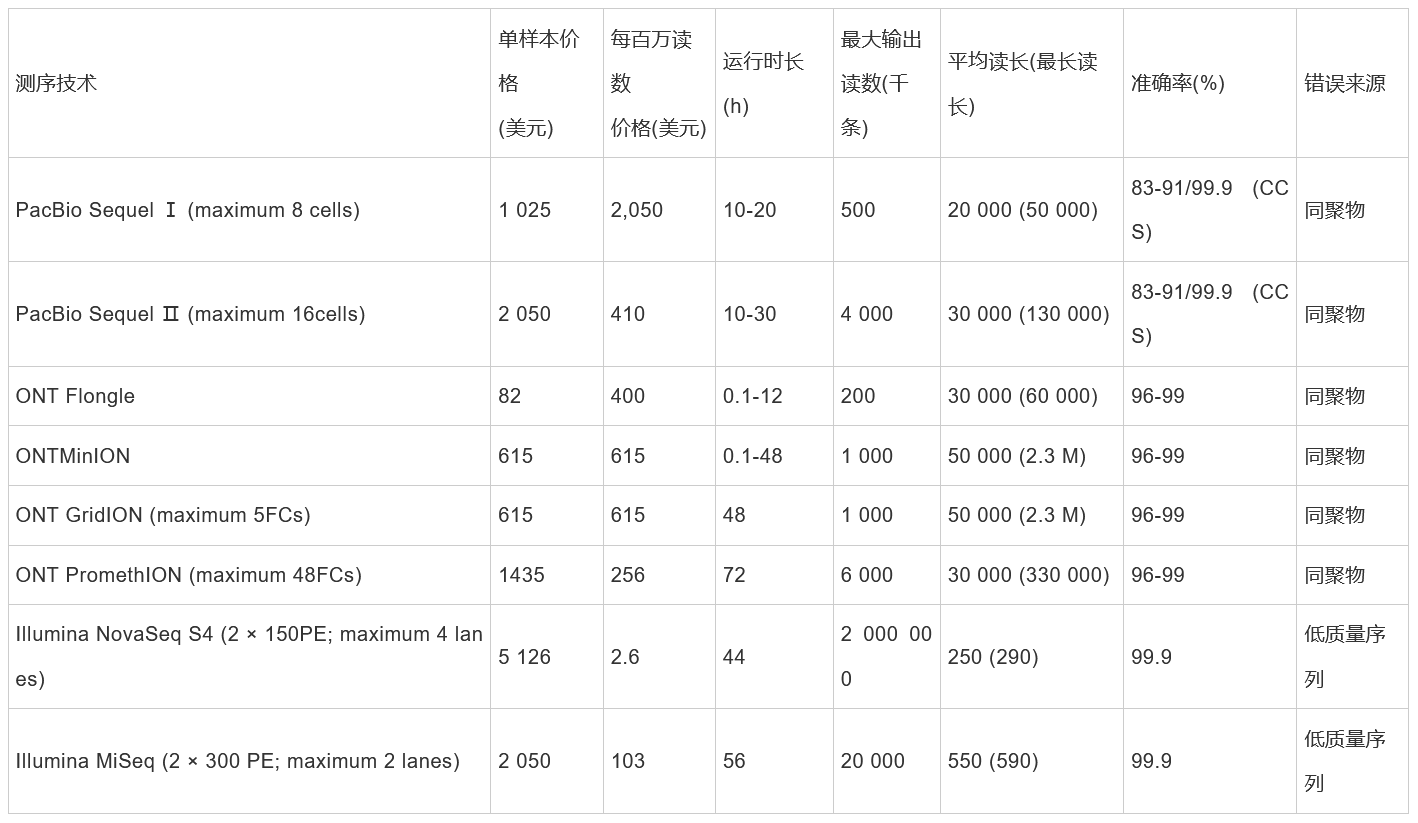 FC: Flow cell; PE: Paired-end; CCS: Circular consensus seque
FC: Flow cell; PE: Paired-end; CCS: Circular consensus seque
2 LRS技术在肿瘤研究中的应用
2.1 基因组变异检测
2.1.1 结构变异检测
结构变异是肿瘤的基因组不稳定性的重要标志,包括易位、大片段缺失、扩增和倒位。结构变异通常是通过驱动易位和扩增使癌基因(Oncogene)激活,驱动缺失和倒位使抑癌基因(tumor suppressor gene, TSG) 失活。因此对肿瘤特异性突变的高灵敏检测,包括小的变异如单碱基替换及大的变异如结构变异,对于指导肿瘤患者早期检测、复发监测和靶向治疗等至关重要。
(1)基于LRS检测癌细胞基因组的结构变异:NGS的诞生为研究基因组微小变异提供了便利工具,但对于较大结构变异如结构变异,NGS技术的短片段(<300 bp)检测仍有很大的局限性。重复区域(包括着丝粒、端粒和其他重复元件)包含人类基因组一半以上的序列(56%),单端测序对于不同位置重复出现的序列片段识别出相同的信息,会导致该序列比对时产生歧义,而LRS(高达20 kb)可以跨越这些重复区域进行片段识别。
LRS针对结构变异检测策略通常分为3种:全面覆盖方法,混合覆盖方法和混合测序方法。
- 全面覆盖方法指所有样本都以中到高覆盖率进行测序;
- 混合覆盖方法指一部分样本以中到高覆盖率进行测序,其余样本使用低覆盖率进行测序;
- 混合测序方法指一部分样本以中到高覆盖率进行测序,其余样本通过短读长测序进行测序。
以上不同策略会影响对群体中常见或罕见变异的检测能力,具体总结为表2。
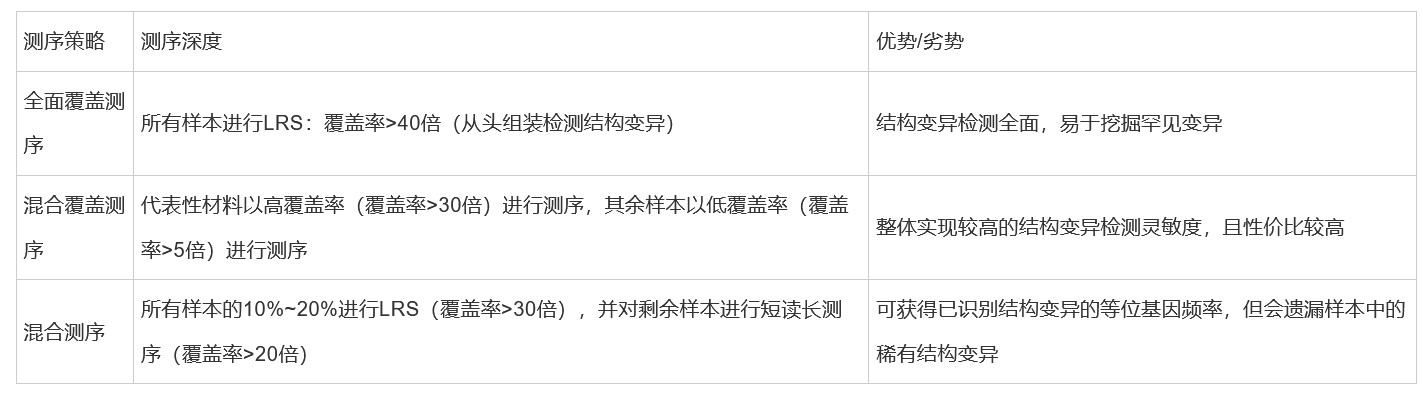 表2 不同策略对结构变异检测能力的影响
表2 不同策略对结构变异检测能力的影响
Hu和Aganezov团队利用ONT和PacBio平台对乳腺癌样本进行了全基因组测序。研究结果表明,即使在相对较低的覆盖率(25X–30X)下,LRS不仅可以检测到单核苷酸变异区域,也可以实现更准确和灵敏的结构变异检测,并且不同LRS方式产出的结果一致率达90%~95%。
此外利用LRS技术检测结构变异在临床应用中也有巨大潜力。研究人员利用MinION技术评估了胰腺导管腺癌细胞系结构变异类型,包括单纯的缺失、易位、倒位以及易位和倒位的组合,该研究对1:100 稀释度的 PCR 扩增产物进行检测,验证了该方法检测患者特异性结构变异的可行性,且每个样本测序深度仅需要500 reads即可快速得到检测结果。
(2)基于LRS的基因组结构变异快速鉴定可用于癌症监测:循环肿瘤DNA(circulating tumor DNA, ctDNA)来源于肿瘤细胞,已被证明与肿瘤负荷呈正线性相关,且在癌症复发出现临床症状前几个月即可检出改变[20]。液体活检可用于检测体液(如血液)中的肿瘤成分,是一种对患者损伤小、可接受度高的肿瘤负荷动态监测方式。在液体活检中检测ctDNA中代表性结构变异,可用于高灵敏度检测和量化ctDNA,因此,准确及时地检测血液中包括结构变异在内的肿瘤相关改变对于患者从早期预警到监测复发均具有重要作用。鉴于测序速度快、体积小和成本低等优点,LRS可能成为早诊、复发或疗效监测过程中癌症相关结构变异检测的理想方式。
为了能够快速和高效地识别一组特异性体细胞结构变异,用于ctDNA监测,Valle-Inclan等利用纳米孔测序的长读长和快速测序能力,开发出一种能够准确选择纳米孔测序数据体细胞SVs模型算法,仅用3天时间即可检测卵巢癌和前列腺癌患者体细胞结构变异,同时,应用该方法DNA样本需求量可以进一步减少到80 ng。此外,研究报道了纳米孔测序技术在同一天内完成了对中枢神经系统肿瘤基因组结构变异、拷贝数及甲基化的检测,并在短时间内进行了IDH1、IDH2和H3F3A的单核苷酸变异分析。Zhang等人[利用SMART测序技术检测ctDNA,追踪未经治疗的中高危/高危弥漫性大B细胞淋巴瘤(diffuse large b-cell lymphoma, DLBCL)的病情演变,对38例中高危/高危DLBCL患者的17个肿瘤活检样本和38个血浆样本进行了基线评估。测序结果显示ctDNA与匹配肿瘤组织之间的突变谱具有良好的一致性。此外,研究人员在ctDNA样本中检测到更多的突变位点,并认为治疗前TP53或B2M突变均提示预后不良。通过长度长测序技术对血液样本的动态分析,证实了其在实时评估疗效方面的应用潜力,以及用于癌症的多模式和快速分子诊断的可行性。
2.1.2 拷贝数变化检测
拷贝数变异是人类基因组变异的一种常见形式,已被证实与许多病理变化相关,包括罕见的基因组疾病、神经系统疾病和癌症。在癌症中,体细胞拷贝数变化(copy number alteration,CNA)促进了基因组的变异,具有诊断、预后和治疗意义。CNA信息可通过染色体分析、核型分析及DNA-FISH[30]等方法检测,然而,这些方法均存在灵敏度或准确度不高的局限性。NGS从短读长测序数据中显示高准确度CNA信息来提高灵敏度和准确度,但此类方法成本较高,应用场所局限于大型、资源丰富的临床中心。
Martignano等利用低覆盖率纳米孔测序检测来自癌症患者血浆核酸分子的拷贝数变化,仅用200万读数就完成了对6名肺癌患者和4名健康受试者的全基因组分子核型检测,并发现了常见的与肺癌相关的拷贝数改变。这是纳米孔测序成功应用于血浆DNA拷贝数分析的案例,展现出了LRS对患者高效、无创检测的潜力。
2.1.3 基因融合检测
基因融合是由于染色体倒位、串联重复、区段缺失或易位事件造成不同的、独立的基因或基因片段发生合并的过程,在癌症中较为常见。例如,慢性髓性白血病中可检出ABL-BCR基因融合,导致费城染色体(Philadelphia chromosome, Ph)的形成;肺腺癌患者的肿瘤组织中EML4-ALK融合基因编码的融合蛋白具有致癌作用。因此检测融合基因,阐明其形成的机制和演化过程对于肿瘤研究具有重要价值。免疫组织化学和荧光原位杂交是临床检测融合基因使用的两种主要方法。以上方法用于分析单个基因,且对于组织样本有一定的要求,在临床实践中可行性低且耗时。因此,LRS技术作为能够以最少的样本量测试多个基因的新技术,可助力融合基因的快速发现。
Jeck William等利用Oxford Nanopore MinION 测序系统,通过修改锚定多重PCR方法进行文库构建,在测序15 min内获得>100条reads,可实现 BCR-ABL1 融合转录本的检测。在Romagnoli等的研究中,LRS技术能在60 h内鉴定出嗜酸性粒细胞增多症患者循环血DNA样本中PDGFRα/β、FGFR1和JAK2重排,该结果与诊断时独立进行的FISH分析完全一致。同时,测序结果为识别这几名患者基因融合位点提供了理论依据,这也是传统方法无法提供的信息。
2.2 全长RNA转录本检测:转录本异构体和长链非编码RNA识别
一些转录亚型的异常表达可能会激活癌基因或使抑癌基因失活,从而导致癌症发生[39]。在RNA水平上研究癌细胞的分子特征,很大程度上依赖于参考转录组的精确度和完整性。然而,由于传统 Illumina 短读长(-150 bp)的限制,通常无法捕捉到转录本中的所有外显子,导致一些新的转录亚型无法识别。Fang等对3例肝细胞癌(hepatocellular carcinoma, HCC)患者的肿瘤组织、门静脉癌栓和癌旁组织进行了纳米孔转录组RNA测序。研究发现,短读长的转录本组装可能会出现许多错误,而长读长在识别结构复杂的长片段新转录本方面具有明显的优势。此外,其他研究也使用LRS成功地发现了细胞中大量新的转录本。以上研究表明LRS是准确组装转录本的有效工具,并且相较于NGS技术能够识别更多未知的转录本。
长链非编码RNA(Long non-coding RNA, lncRNA)参与基因组修饰、转录激活、转录干扰、核内运输等过程,其在肿瘤发生发展过程中的调控作用已被大量研究证实。lncRNA会因细胞类型、组织类型和物种类型的不同产生多种同工型。RNA-seq可揭示lncRNA表达差异,但表达差异不足以代表其作用方式,因此LRS技术为探索lncRNA不同加工方式及生物学功能提供了有力工具。为了加速lncRNA注释,GENCODE联盟开发了RNA捕获长测序(RNA capture long seq, CLS),它将靶向 RNA 捕获与第三代LRS相结合,所生成的全长转录本模型能够明确描述lncRNA的基因组特征,包括启动子和基因结构,以及蛋白质编码潜能。
2.3 表观遗传修饰识别
表观遗传学在驱动癌症及其异质性方面具有重要作用,基因表达的表观遗传调控主要依赖于DNA甲基化和组蛋白修饰。迄今为止,研究DNA甲基化通常先利用亚硫酸氢盐处理,将胞嘧啶转化为尿嘧啶进而将表观遗传信息转化为遗传信息,再进行大规模平行DNA测序。然而,基于亚硫酸氢盐处理的测序无法区分胞嘧啶、5-甲基胞嘧啶 (mC)和5-羟甲基胞嘧啶 (hmC),这增加了文库制备的复杂性以及来自不完全化学转化等偏差的可能性。此外,常用的NGS技术存在读取长度短的限制,使得一些区域难以定位,无法进行等位基因特异性甲基化的研究。
Rand等和 Simpson等首次使用纳米孔测序实现了全基因组DNA 甲基化分析。Ewing等人利用ONT测序技术实现了转座元件表观基因组的全面分析,同时评估转座因子(transposable elements)甲基化和检测体细胞转座因子动员。Davenport等和Zhang等使用纳米孔测序技术在HCC全基因组范围内绘制mC信号以识别新的TSG。该研究根据再生肝脏和原发性HCC的基因转录谱与甲基化数据的整合识别出10个潜在的TSG,随后对其中一种候选物——葡萄糖激酶的作用进行了探究,验证其通过诱导细胞内乳酸积累来抑制HCC细胞的增殖,参与HCC的发生发展。此后,研究者们开发及改进了基于LRS技术对表观遗传景观进行建模和分析的工具[55,56],将现有DNA甲基化检测技术向前推进了一步。
3 总结与展望
LRS技术助力人类癌细胞基因组及全转录组图谱绘制,使肿瘤患者游离DNA检测、大片段结构变异和甲基化等快速检测成为可能,为临床快速筛选、疾病诊断及预后判断提供了有力依据。LRS技术在肿瘤研究中的应用正在不断扩展,特别是在缺少参考基因组信息的情况下,能够得出更为准确的拼接信息。例如,研究者将宫颈癌多组学数据与人乳头瘤病毒整合位点LRS数据相结合,识别和筛选出新的宫颈癌候选靶基因(BNC1、RSBN1、USP36 和 TAOK3),并在细胞迁移、增殖和集落形成等体外功能研究中验证了其潜在致癌作用。
LRS技术尚在起步阶段,仍然存在一些尚待解决的问题。首要挑战则是对最低样本量的需求。LRS技术相对NGS对检测DNA或RNA的质量和数量要求更低,但仍需要微克级DNA来制备用于全基因组测序的文库,特别是临床血液及体液样本提取的DNA量通常很有限,因此,LRS应用于临床还需要进一步减少对样本量的需求。另一个挑战则是ONT和PacBio测序存在较高的原始错误率以及长扩增子嵌合体形成率,有待于通过升级优化检测平台和下游分析流程规避可能的错误。例如,ONT发布的PromethION流通池R10.3版本测序准确度远高于之前版本,在单读时测序准确性可达到99%,有助于改善当前长读测序的碱基准确度低的问题,进一步帮助改进下游分析。此外,针对LRS数据的下游分析如重复序列比对、基因组结构变异检测和甲基化分析等,依赖人工智能算法、软件和数据库等生物信息学工具。目前已有的针对NGS数据开发的算法无法解决LRS数据高原始错误率的问题,因此开发更适合处理长序列片段数据的算法,有助于LRS技术准确度的提升。
LRS技术在肿瘤研究中的应用思路和探索领域正在不断扩展,特别是针对一些具有高突变率的肿瘤,如血液肿瘤中的各类白血病、多发性骨髓瘤及恶性淋巴瘤等,其可通过以下应用方向发挥重要作用:
- 检测肿瘤早期发生的基因突变,对癌症进行早期预警;
- 根据分子标志物对疾病进行鉴别诊断及分子分型;
- 根据患者的基因变异选择合适的靶向药,做到精准治疗;
- 利用基因变异导致患者对化疗药物耐受性不同,进行化疗副作用预测;
- 利用LRS技术检测微小残留灶(MRD)进行疗效判断及复发监测,从而进一步提高分子诊断在肿瘤疾病的早期诊断、精准治疗、判断疗效、复发预测等全程管理中的重要应用价值。
尽管该技术仍然存在诸多亟待解决的问题,LRS仍是测序技术不断更新换代中的重要里程碑。相信随着技术的革新,未来LRS技术将加速肿瘤基因组学及表观遗传组学研究,窥探既往难以发现的分子生物学信息,为新的肿瘤生物标志物识别及诊疗方式开发提供新的思路。
文献来源
杨谨衔,陈淑桢,王红阳,文文.长读长测序技术在肿瘤领域中的应用:进展与挑战[J].海军军医大学学报:1-9.
- 本文固定链接: https://maimengkong.com/kyjc/1977.html
- 转载请注明: : 萌小白 2025年7月22日 于 卖萌控的博客 发表
- 百度已收录
