我们拿到测序的数据后,首先根据barcode区分每个样本,然后进行生信分析。这里先抛个砖,简单说说生信分析,下面以细菌为例说明我们的分析结果。
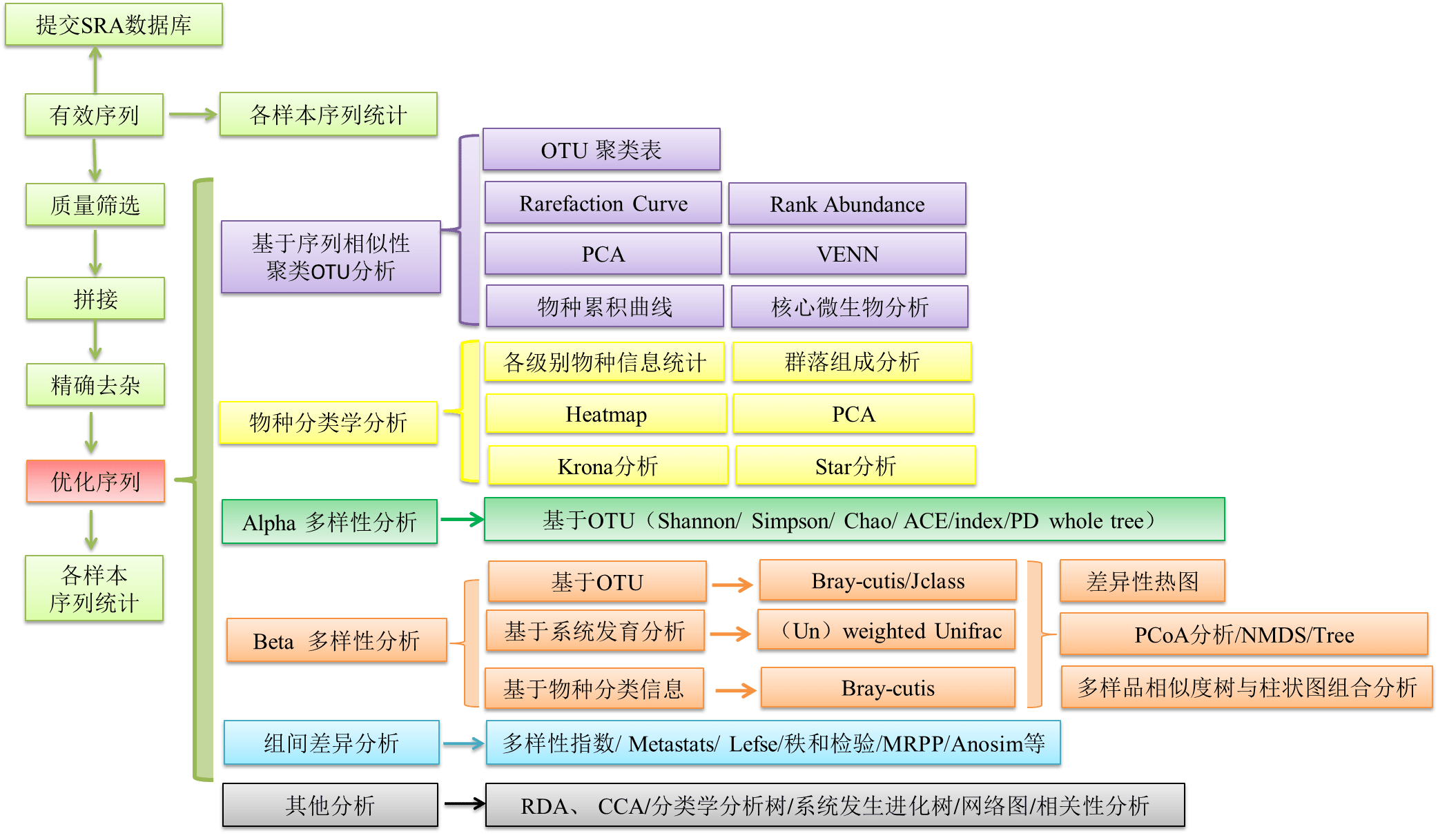
生物信息分析流程
主要分为6大步:
n 序列优化
n 优化序列OTU聚类及注释
n OTU分析及注释物种分析
n 基于alpha多样性分析
n 基于beta多样性分析
n 差异分析
详细流程图如下:
一、序列优化
• 1、原始数据样品区分统计
目前高通量测序基本都是采用多个样本平行测序,为了区分每个样本,建库时引入一段标识序列--barcode标签序列。测序返回数据后根据barcode标签将各样本拆分开并以fastq格式保存,每个样本都有fq1/fq2两个文件,序列按顺序一一对应。后续生信分析及上传NCBI数据库SRA都使用该数据。
• 2、有效序列质控及拼接
a) 使用Trimmomatic软件按划框法截取低质量序列
b) 使用FLASH软件根据PEreads之间的overlap关系,将reads拼接。
• 3、序列优化
对拼接好的序列进行质控过滤。因PCR扩增会引入非特异性扩增及模糊碱基/单碱基高重复区,还有一部分嵌合体。
a) 特异引物去除非特异性扩增
b) 去除模糊碱基/单碱基高重复区及不符合长度的序列
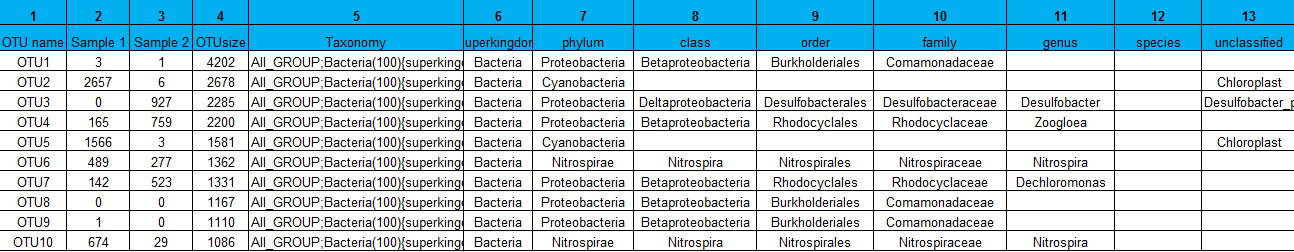
二、OTU聚类及物种信息注释统计
1. 使用usearch中的cluster_otus命令聚类OTU
2. 使用usearch中的uchime_ref命令及嵌合体数据库去除PCR引入的嵌合体
3. mothur命令classify.seqs跟silva128数据库比对,进行物种注释
物种注释结果按门纲目科属种提取,OTU分布及物种信息分别均一化到100%进行统计分析。
三、OTU分析及注释物种分析
1、基于OTU层面分析
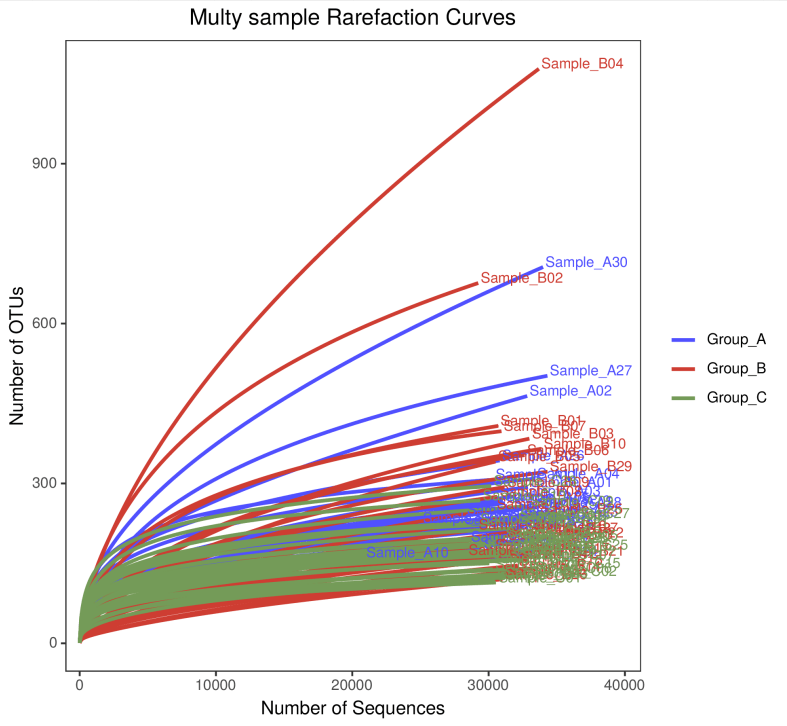
1.1 稀释性曲线与物种累计曲线
在单样本中,稀释性曲线是从样本中随机抽取一定数量的reads,统计这些reads所代表的物种数目,并以reads数与物种数来构建曲线。单样本中,稀释曲线可以用来表示测序深度情况;多样本中,对于不同测序数据量,可以用来比较物种丰富度。曲线越趋平缓,说明测序深度合理。
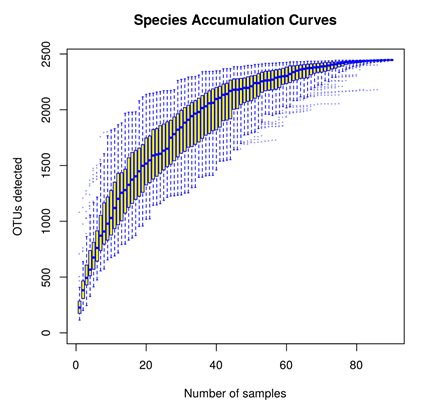
与稀释性曲线相对的,基于所有样本,物种累积曲线(SpeciesAccumulationCurves)用于描述随着抽样量的加大物种增加的状况,是理解调查样地物种组成和预测物种丰富度的有效工具。曲线趋于平缓则表示抽样充分。表示了随着样本数的增加而产生的OTU(物种)数目。
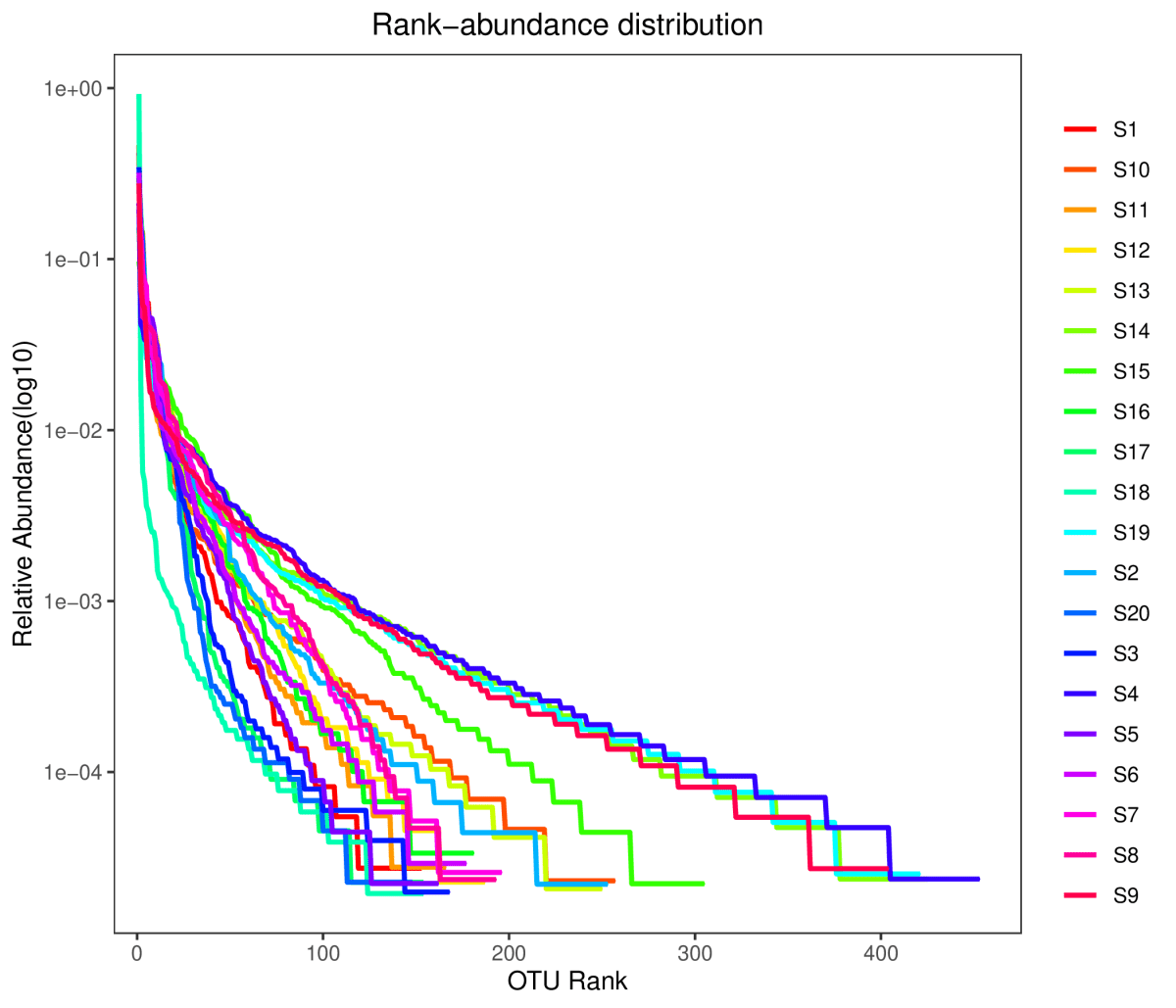
1.2 丰度等级图—RankAbundance
RankAbundance曲线是在样本中按OTU丰度从大到小排序后做的曲线图。即展示物种丰度又展示了物种均匀度。
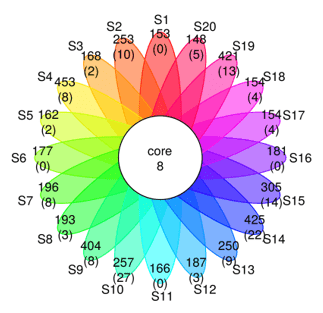
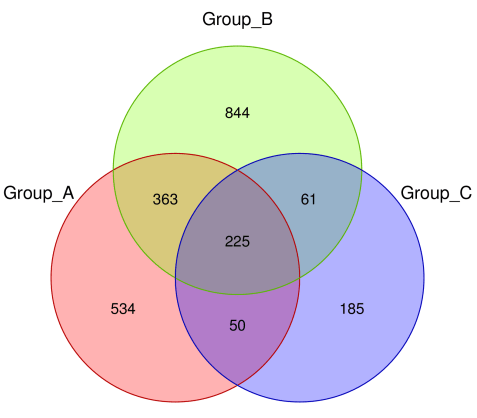
1.3 维恩图/花瓣图
VENN图/花瓣图都是用于统计多个样本(或分组)中共有/独有OTU数数目的图。
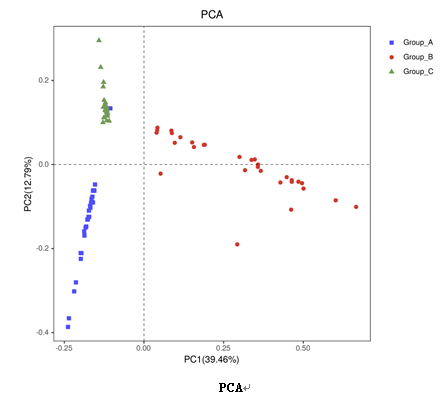
1.4 主成分分析PCA
PCA(PrincipalComponentAnalysis),即主成分分析,是一种对数据进行简化分析的技术,这种方法可以有效的找出数据中最“主要”的元素和结构,去除噪音和冗余,将原有的复杂数据降维,揭示隐藏在复杂数据背后的简单结构。样品组成越相似,反映在PCA图中的距离越近。
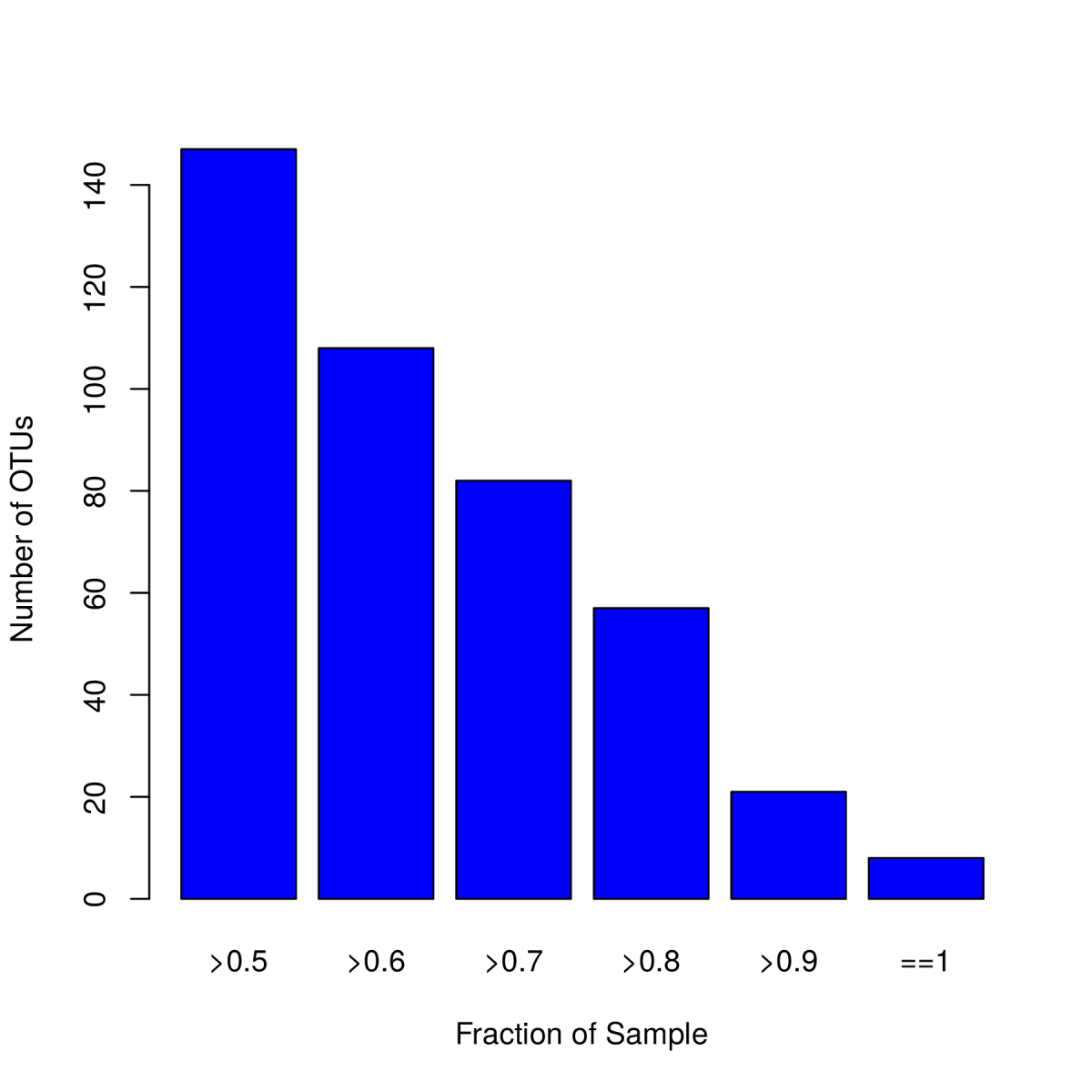
1.5 核心微生物分析
核心微生物分析即Coremicrobiome分析。表示某个OTU及其所代表的的物种在所有样本中存在。
2、基于物种层面分析
2.1 热图Heatmap
Heatmap:用颜色变化来反映二维矩阵或表格中的数据信息,很直观地将数据值的大小以定义的颜色深浅表示出来。先对物种/样本间丰度相似性聚类,然后将高低丰度的物种分块聚集。通过颜色梯度及相似程度来反映多个样品在各分类水平上群落组成的相似性和差异性。
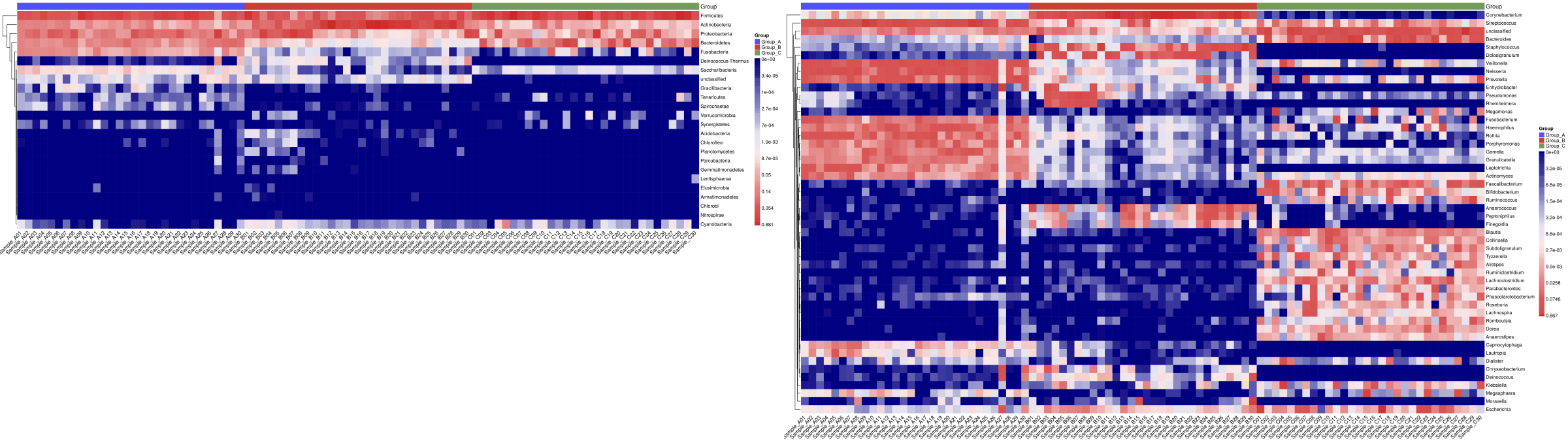
热图(门和属水平)
2.2 群落组成分析
根据分类学分析结果,可以得知一个或多个样品在各分类水平上的分类学比对情况。在结果中,包含了两个信息:
a) 样品中微生物组成成分
b) 样品中各微生物的序列数,即各微生物的相对丰度。
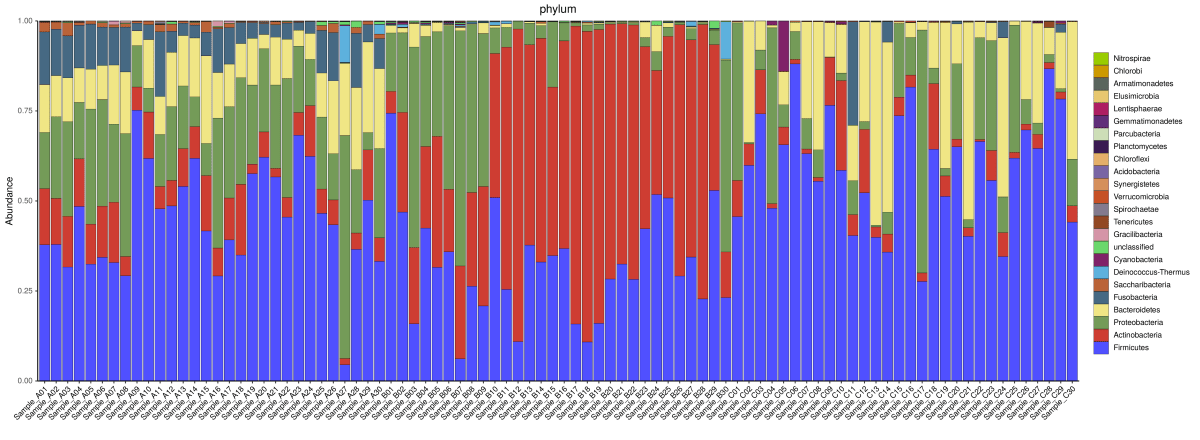
门水平物种分布柱状图
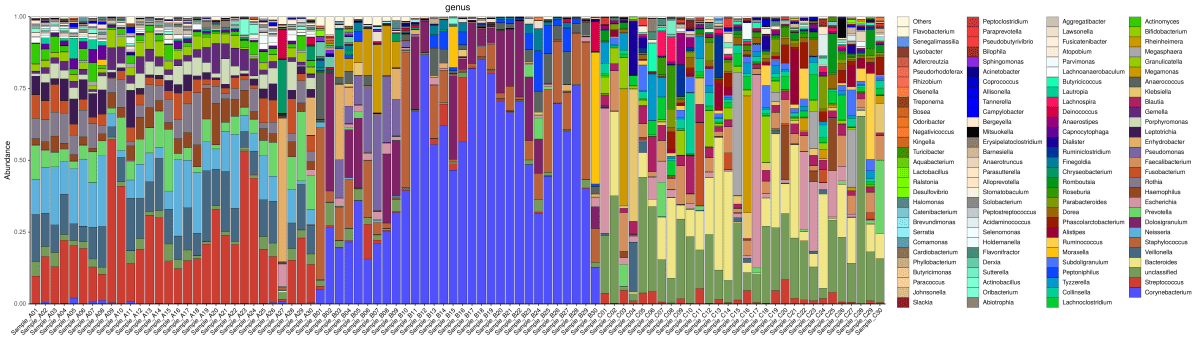
属水平物种分布柱状图
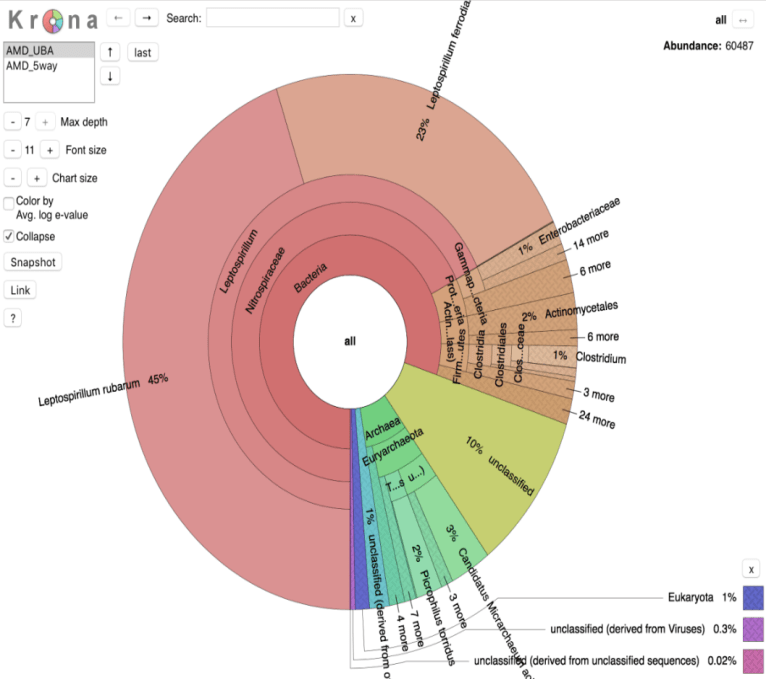
2.3 Krona分析
使用krona软件对物种注释结果进行可视化展示,展示结果中,圆圈从内到外依次代表不同的分类级别,扇形的大小代表不同OTU注释结果的相对比例。
2.4 Star分析
在属分类水平,对丰度最高的前10个属的物种丰度做Star图。一个星形图代表着一个样品的物种相对丰度信息。每个星形图中的扇形代表一个物种,用不同颜色区分,用扇形的半径来代表物种相对丰度的大小,扇形的半径越长表示丰度高,反之丰度相对较低。
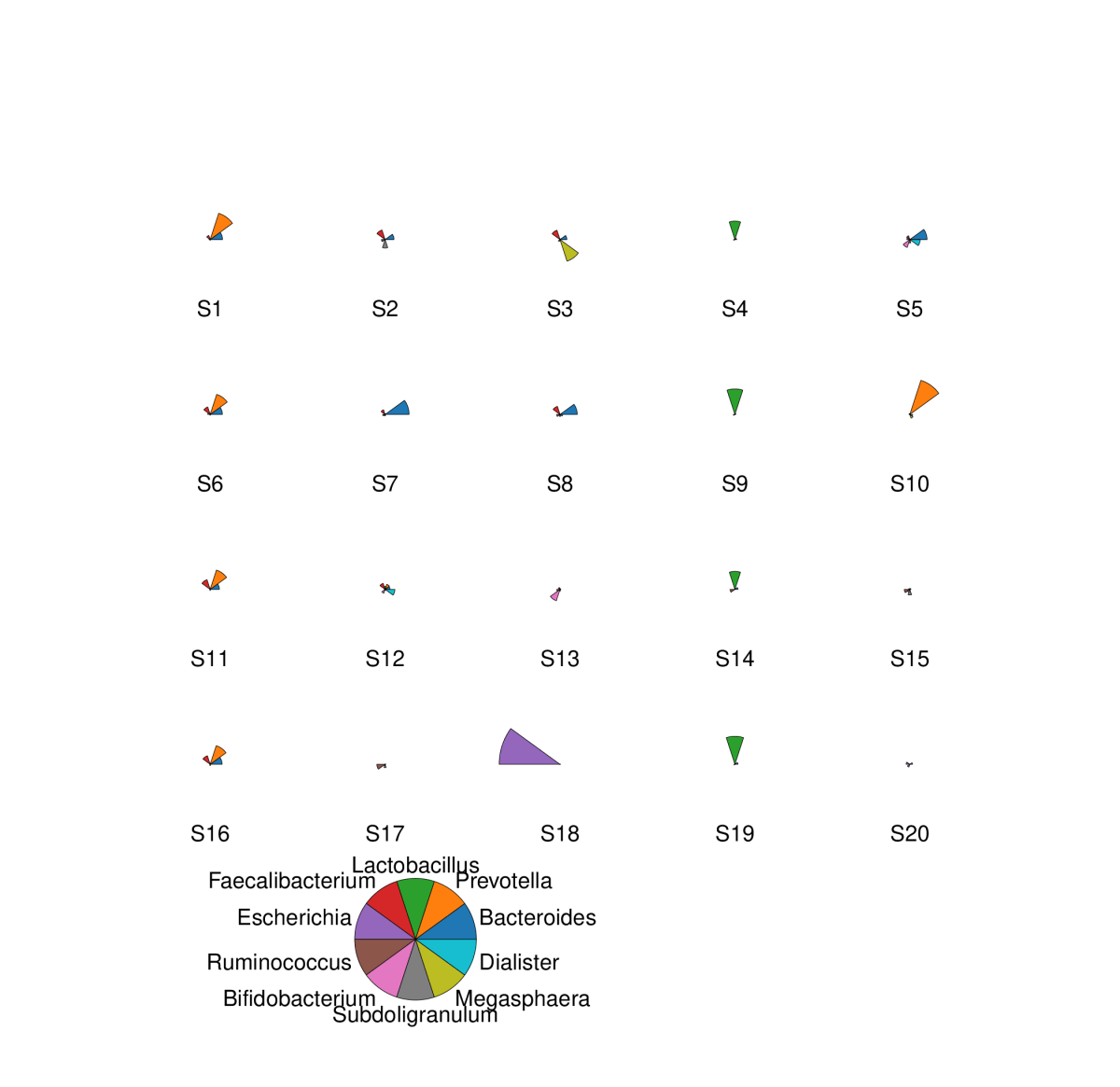
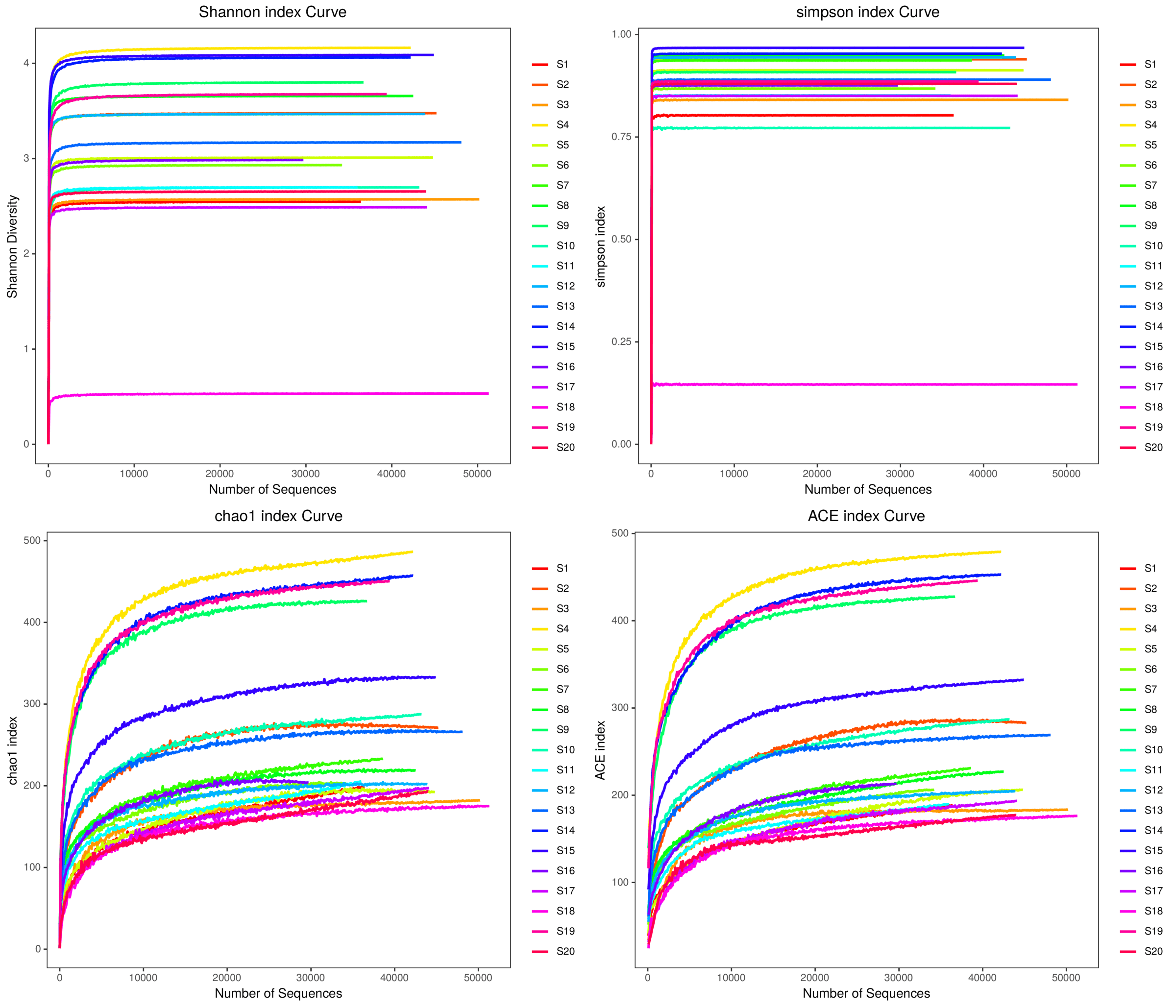
四、Alpha多样性分析
Alpha多样性(Alphadiversity)只计算样本内部而不考虑样本间,包括chao指数、ace指数,shannon指数以及simpson指数,PD_whole_tree指数等。
其中,chao和ACE指数反映样本中群落的丰富度(speciesrichness),只考虑物种数量,不考虑物种丰度。shannon指数及simpson指数反映群落的多样性(speciesdiversity),受到样本群落中的物种丰度度(speciesrichness)和物种均匀度(speciesevenness)的影响。丰富度相同,均匀度越大,认为该群里多样性越大。 PD whole tree指数是基于系统发生树计算的一个多样性,用各个样品中OTUs的代表序列计算出构建系统发生树的距离,将某一样品中的所有代表序列的枝长加和,从而得到的数值。
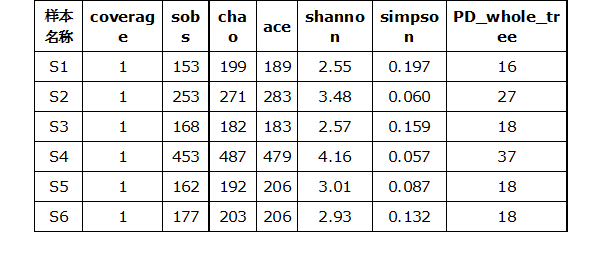
按不同随机抽样OTU数目下计算每个样本的Alpha多样性指数值,做如下图展示
五、Beta多样性分析
1、基于OTU的Beta多样性
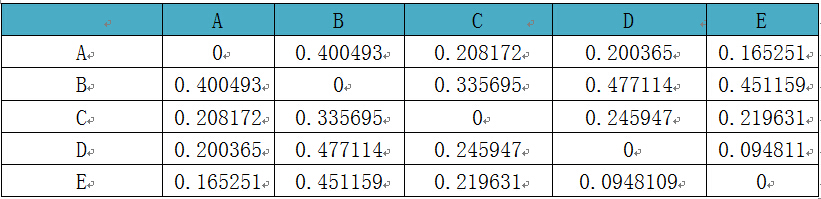
相比较单样本内计算的Alpha多样性,Beta多样性(Betadiversity)则是用来分析样本间的物种多样性方面存在的差异。分析各类群在样品中的含量,进而计算出不同样品间的Beta多样性值。多种指数可以衡量Beta多样性,常用的为Bray-Curtis,Jaccard,weightedunifrac,unweightedunifrac。
a) Jaccard法:该方法通过Jaccard计算样本之间的差异性,分析时只考虑样本中OTU的有无,不考虑丰度
b) Bray-curtis法:该方法通过Bray-curtis计算样本之间的差异性,分析时既考虑样本中OTU的有无,又考虑丰度
2、基于进化关系的Beta多样性
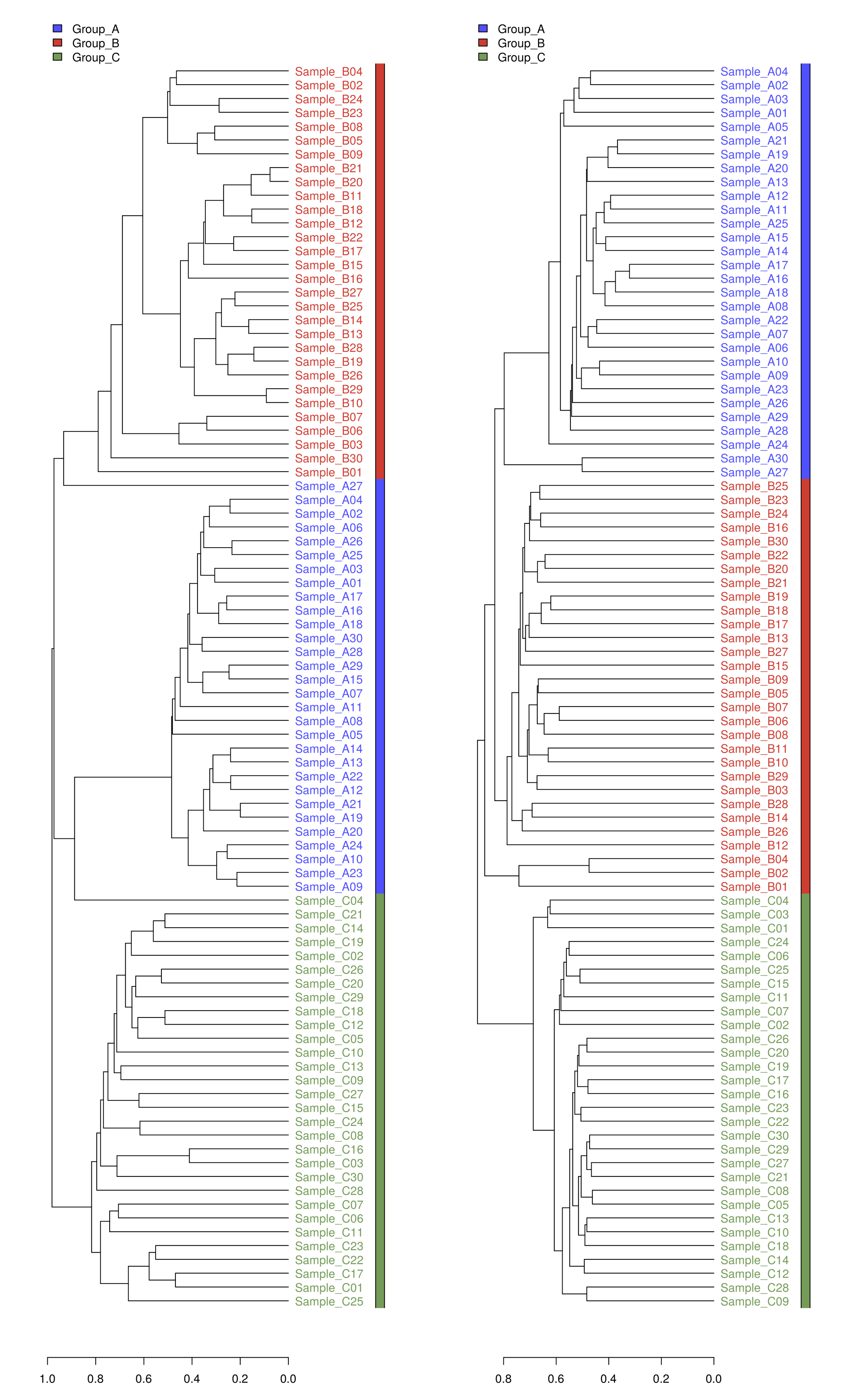
UniFrac分析利用各样品序列间的进化信息来比较环境样品在特定的进化谱系中是否有显著的微生物群落差异。对样品两两之间进行比较分析,得到样品间的unifrac距离矩阵。Unifrac度量标准根据构建的进化树枝的长度计量两个不同环境样品之间的差异,差异通过0-1距离值表示,进化树上最早分化的树枝之间的距离为1,即差异最大;若两个群落完全相同,UniFrac值为0。
a) unweighted unifrac可以看出它只考虑序列是否在群落中出现,而不考虑序列的丰度。
b) Weighted unifrac方法,就是在unweighted unifrac的基础上,将序列的丰度纳入考虑,它能够区分物种丰度的差别。
通过层次聚类(Hierarchicalcluatering)中的非加权组平均法UPGMA构建进化树等图形可视化处理Unifrac分析得到的距离矩阵。
NMDS非度量多维尺度法是一种将多维空间的研究对象(样本或变量)简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法。其特征为将对象间的相似性或相异性数据看成点间距离的单调函数,在保持原始数据次序关系的基础上,用新的相同次序的数据列替换原始数据进行度量型多维尺度分析。
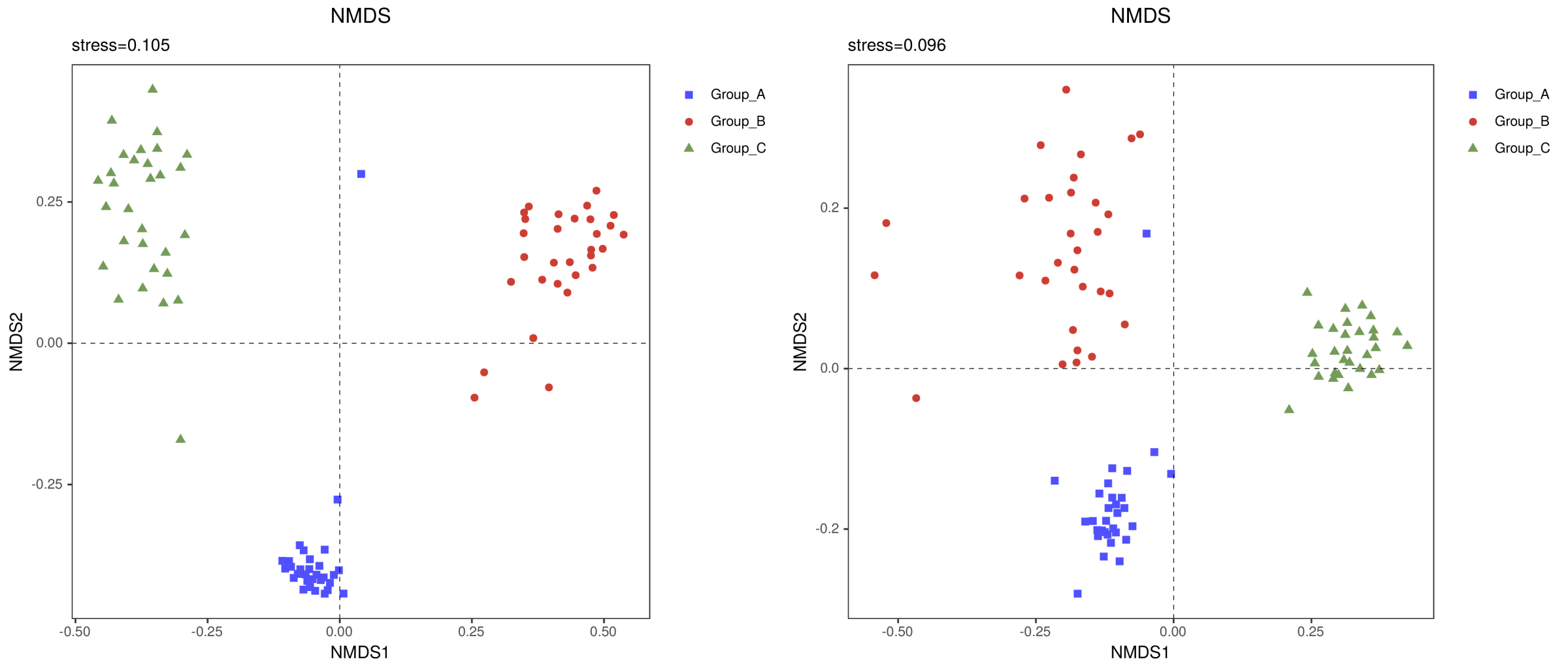
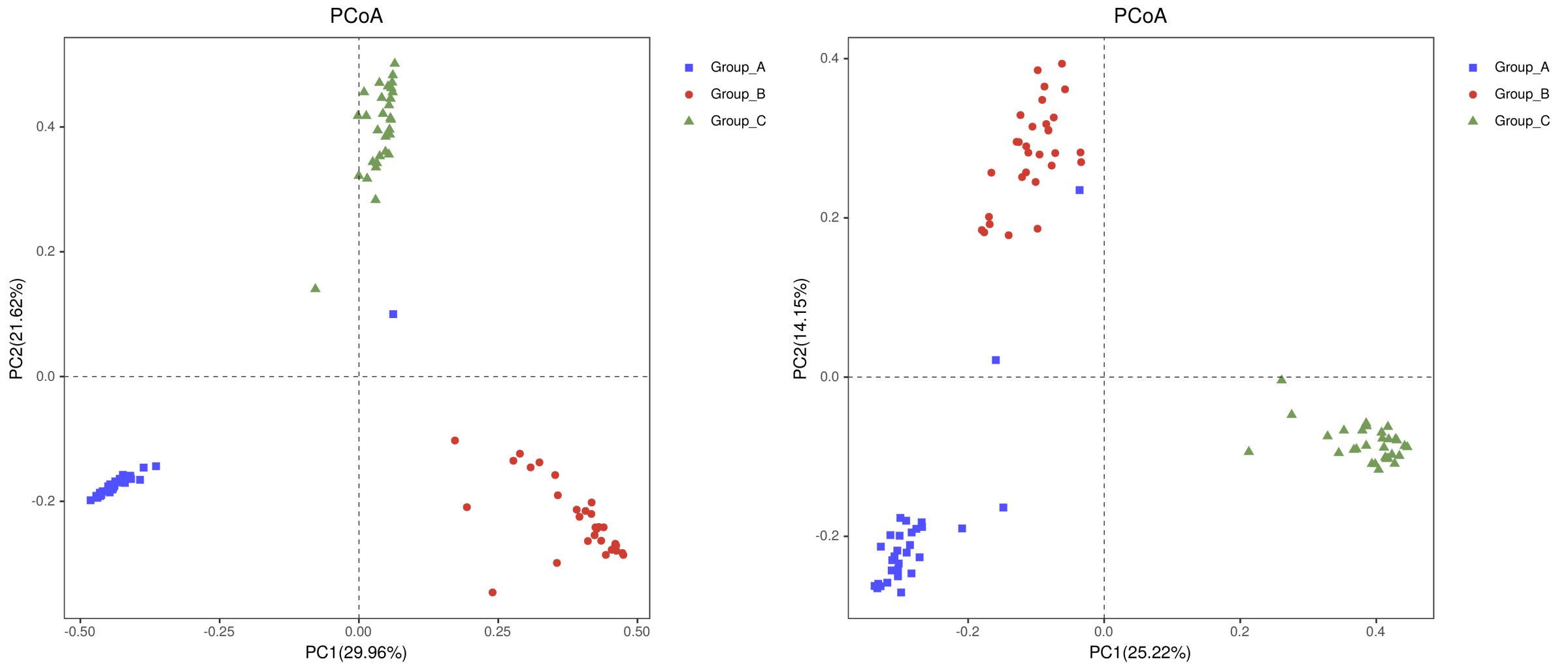
PCoA(principalco-ordinatesanalysis) 是一种研究数据相似性或差异性的可视化方法,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,PCoA可以找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统。通过PCoA可以观察个体或群体间的差异。
六、差异分析
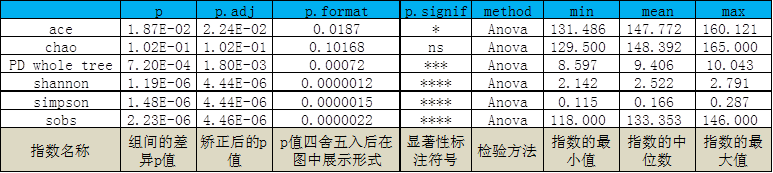
1、Alpha多样性指数差异性分析
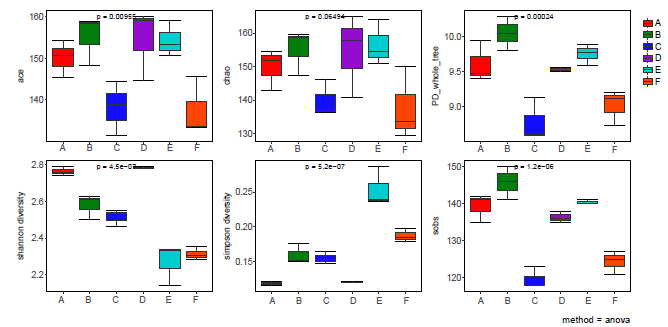
如样品有提供分组信息,且每组样品个数不小于3,将对组间的Alpha多样性指数进行差异分析,包括参数检验和非参数检验。方法:两组样本之间采用方法:wilcox;三组样本或多组样本之间采用方法:krustal;两组样本之间采用方法:Ttest;三组样本或多组样本之间采用方法:anova。以例图说明文件含义:
该结果以箱线图的形式展示出来,纵坐标为各指数的值,组间差异通过p值来展示,两组之间的差异将P值的数字展示在图中,多组之间的差异在图的左上角标注。
所有指数显示在一张图上:
2、ADONIS组间群落显著性差异分析
置换多元方差分析(Permutational multivariate analysis of variance,PERMANOVA),又称非参数多因素方差分析(nonparametric multivariate analysis of variance)、或者ADONIS分析,其本质是基于F统计的方差分析,依据距离矩阵对总方差进行分解的非参数多元方差分析方法。使用PERMANOVA可分析不同分组因素对样品差异的解释度,并使用置换检验进行显著性统计。
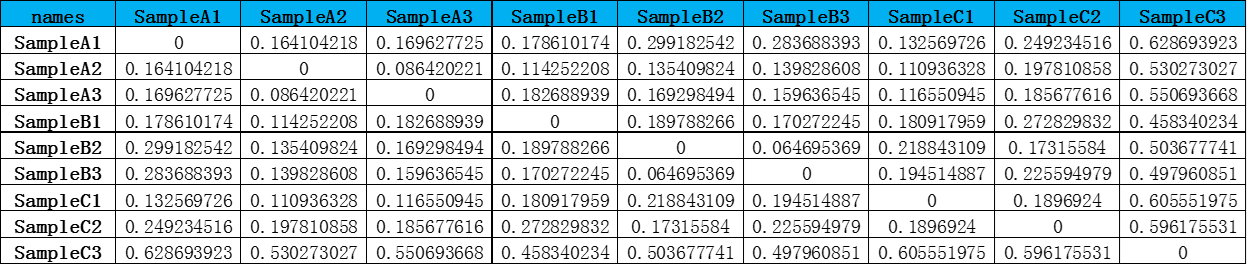
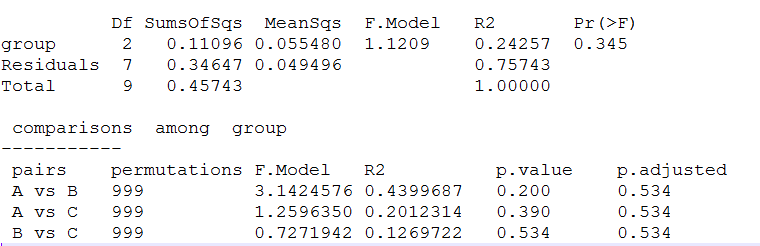
注释:group行即为本示例group分组的组间差异检验结果,Residuals为残差,Total为group和Residuals的加和。对于各项统计内容:Df,自由度,其值=所比较的分组数量-1;SumsOfSqs,即Sums of squares,总方差,又称离差平方和;MeanSqs,即Mean squares,均方(差);F.Model,F检验值;R2,即Variation (R2),方差贡献,表示不同分组对样品差异的解释度,即分组方差与总方差的比值,R2越大表示分组对差异的解释度越高;Pr (>F),显著性p值,默认p<0.05即存在显著差异。
3 Anosim分析(每组样品数≥5)
相似性分析Anosim分析是一种非参数检验,用来检验组间(两组或多组)的差异是否显著大于组内差异,从而判断分组是否有意义。如果R值大于0,说明组间的差异大于组内差异,分组较为合理,反之R值若小于0,则说明组间差异小于组内差异,分组欠妥。P值越小组间差异越大。
例图:
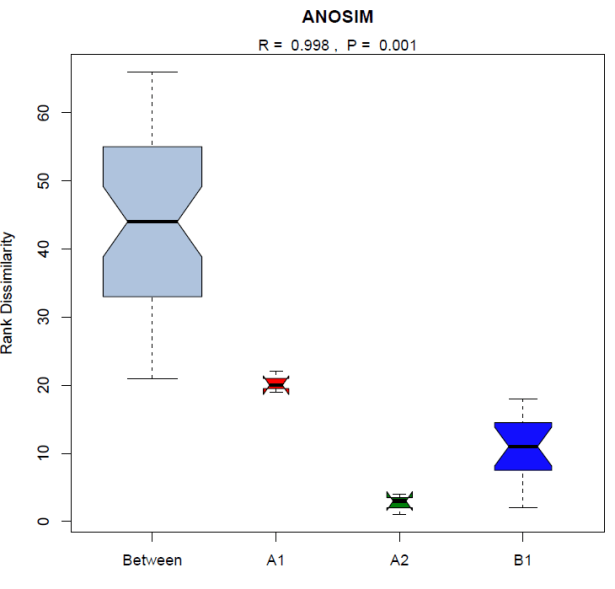
注:
横坐标表示所有样品(Between)以及每个分组(A1、A2、B1),
纵坐标表示unifrac距离的秩。
Between组相对于其他每个分组的秩较高时,则表明组间差异大于组内差异。
R介于(-1,1)之间,R大于0,说明组间差异显著;
R小于0,说明组内差异大于组间差异,统计分析的可信度用P表示,P小于0.05表示统计具有显著性。
4 MRPP分析
MRPP组间差异分析是用于分析组间微生物群落结构的差异是否显著的一种分析方法,通常与PCoA、NMDS等降维图配合使用。
下表为基于进化关系分析的结果:
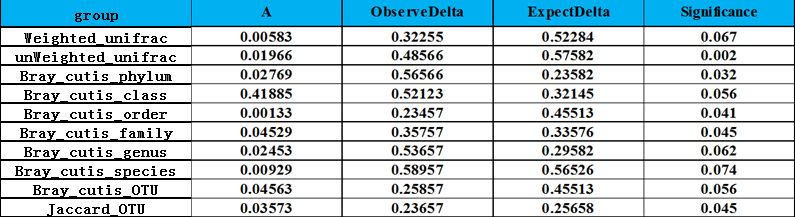
注:Group:分为weighted_unifrac、unweighted_unifrac等;
A:A统计量;
ObserveDelta:weighted_unifrac、unweighted_unifrac等方法计算距离指数对应的ObserveDelta值;ExpectDelta:weighted_unifrac、unweighted_unifrac等方法计算距离指数对应的ExpectDelta值;
Significance:weighted_unifrac、unweighted_unifrac等方法计算距离指数对应的显著性p值;
A值大于0说明组间差异大于组内差异,A值小于0说明组内差异大于组间差异;ObserveDelta值越小说明组内差异越小;ExpectDelta值越小说明组间差异越小;
5 各分类级别物种差异分析
采用参数检验和非参数检验两种方法进行组间的差异分析
非参数检验:两组样本之间采用方法:wilcox;三组样本或多组样本之间采用方法:krustal;
参数检验:两组样本之间采用方法:T-test;三组样本或多组样本之间采用方法:anova.。
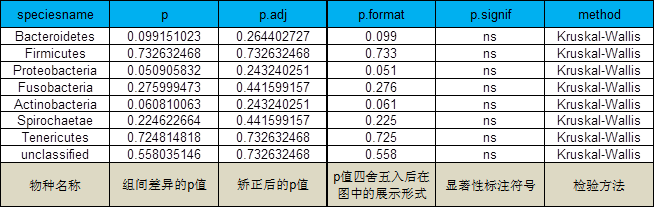
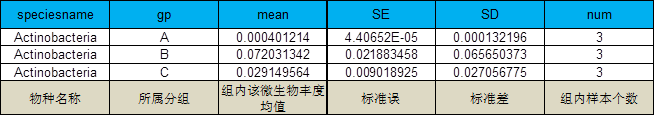
例图:
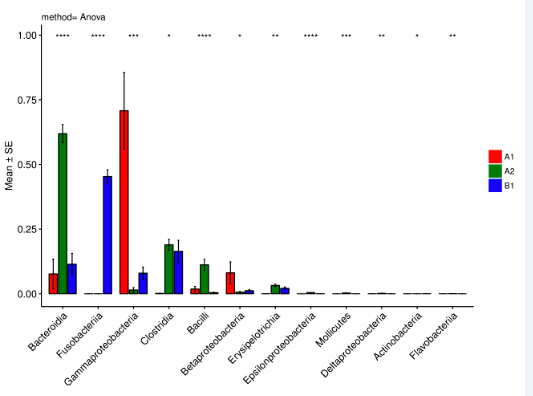
注:选择p<0.05的物种绘制柱形图,每个柱子为差异物种的相对丰度,误差线为标准误,其中﹡代表0.01<p<0.05,﹡﹡代表0.001<p<0.01,﹡﹡﹡代表0.0001<p<0.001,﹡﹡﹡﹡代表p<0.0001。
6 RDA/CCA分析
RDA或者CCA是基于对应分析发展而来的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。此分析是主要用来反映菌群与环境因子之间关系。RDA是基于线性模型,CCA是基于单峰模型。分析可以检测环境因子、样品、菌群三者之间的关系或者两两之间的关系。
例图:
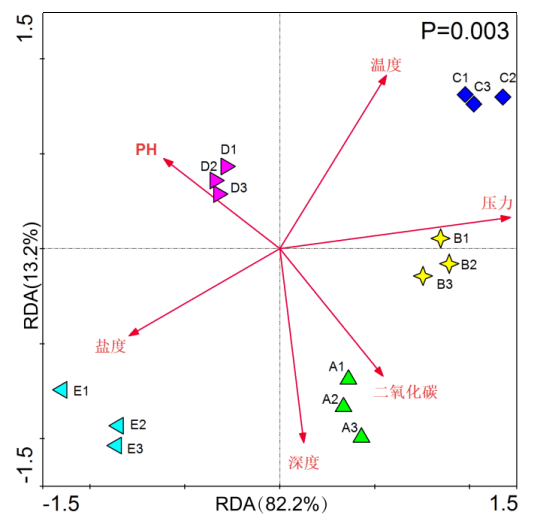
注:图中数字表示样品名,不同颜色或形状表示不同环境或条件下的样本组;箭头表示环境因子;物种与环境因子之间的夹角代表物种与环境因子间的正、负相关关系(锐角:正相关;钝角:负相关;直角:无相关性);由不同的样本向各环境因子做垂线,投影点越相近说明样本间该环境因子属性值越相似,即环境因子对样品的影响程度相当。
7 LEfSe组间群落差异分析
LEfSe(LDAEffectSize)是一种用于发现高维生物标识和揭示基因组特征的软件。包括基因,代谢和分类,用于区别两个或两个以上生物条件(或者是类群)。该算法强调的是统计意义和生物相关性。让研究人员能够识别不同丰度的特征以及相关联的类别。
LEfSe通过生物学统计差异使其具有强大的识别功能。然后,它执行额外的测试,以评估这些差异是否符合预期的生物学行为。具体来说,首先使用non-parametric factorial Kruskal-Wallis(KW) sum-rank test(非参数因子克鲁斯卡尔—沃利斯和秩验检)检测具有显著丰度差异特征,并找到与丰度有显著性差异的类群。最后,LEfSe采用线性判别分析(LDA)来估算每个组分(物种)丰度对差异效果影响的大小。
例图:
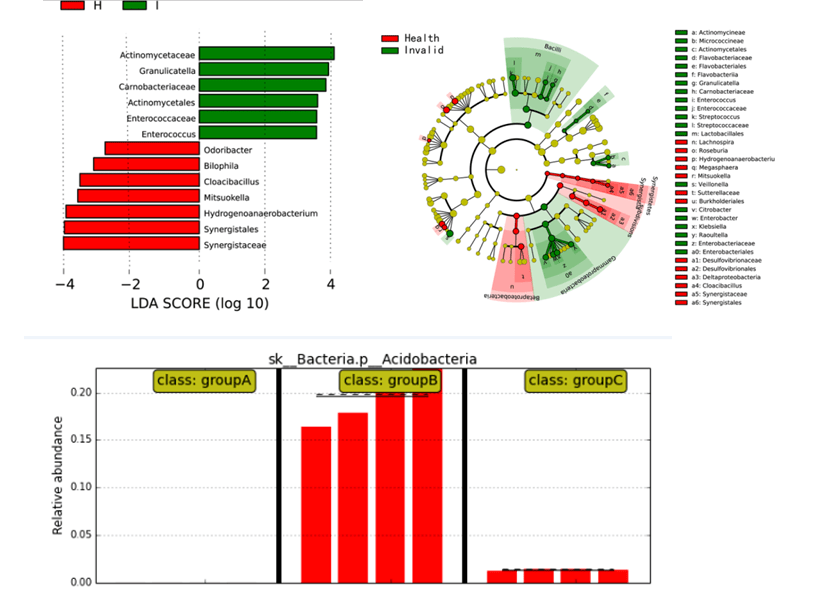
注:左上图为统计两个组别当中有显著作用的微生物类群通过LDA分析(线性回归分析)后获得的LDA分值。右上图为聚类树,红色区域和绿色区域表示不同分组,树枝中红色节点表示在红色组别中起到重要作用的微生物类群,绿色节点表示在绿色组别中起到重要作用的微生物类群,黄色节点表示的是在两组中均没有起到重要作用的微生物类群。图中英文字母表示的物种名称在右侧图例中进行展示。这个由内至外辐射的圆圈图为聚类树,依次代表了由门、纲、目、科、属、(或种)的分类级别。在不同圆圈层上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径与该分类的相对丰度大小呈正比。
下面图差异物种通过柱形图来显示,图最上面的注释为物种信息,纵坐标为相对丰度,横坐标为样本名称(每个柱子为一个样本,图中只标示分组名称,不标示样本名称),不同的分组用黄色框显示。此图中,物种丰度最高,说明该物种是本分组的biomarker。
生成结果文件的说明:
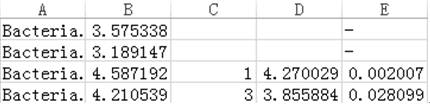
注:第A列biomarker名称;
第B列是平均丰度最大的log10的值,如果平均丰度小于1的按照1来计算;
第C列是差异基因或物种富集的组名称;
第D列是LDA值;
第E列是Kruskal-Wallis秩和检验的p值,如果不是biomarker则用“-”表示。
- 本文固定链接: https://maimengkong.com/kyjc/1232.html
- 转载请注明: : 萌小白 2022年10月4日 于 卖萌控的博客 发表
- 百度已收录
