前段时间为大家介绍过绝对定量转录组测序可以为研究工作带来的改善,你还记的吗?
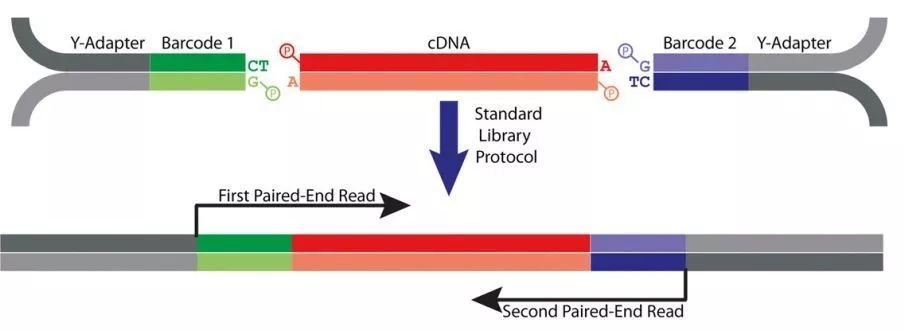
图1 绝对定量转录组测序建库原理
绝对定量转录组测序是这样做的:使用含有UMI标签(或Barcode)的接头与cDNA连接,然后进行PCR文库扩增和上机测序(图1)。
定量更准确
由于每个cDNA扩增产物都带有unique的UMI标签,在数据分析时,通过识别UMI标签,就可以容易地识别哪些序列是PCR扩增产生的(相同的UMI),而哪些序列的多拷贝是真实的(不同的UMI),因而,相比常规转录组测序,转录本定量的准确性大幅提高(图2)。
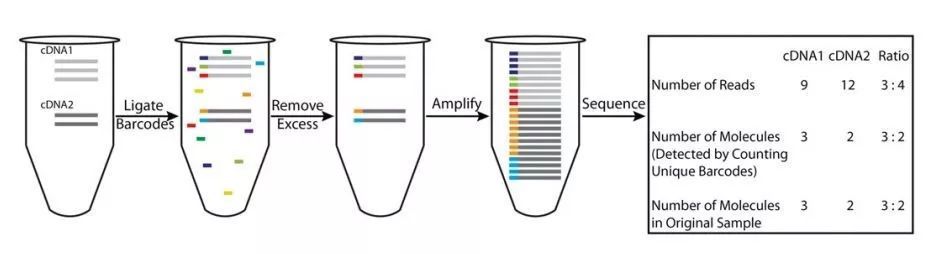
图2 绝对定量转录组测序定量原理
序列测定更准
绝对定量转录组测序不仅定量更准确,而且序列测定的准确性也显著提高,更适合转录本SNP的鉴定,这是因为带有相同UMI标签的相同序列会进行序列校正,即去除掉因扩增或测序错误产生的假突变(图3)。
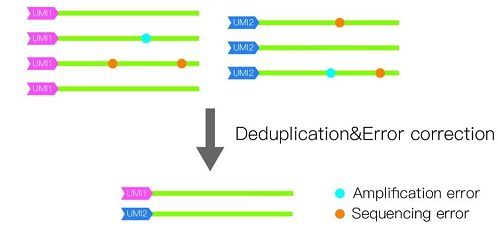
图3 绝对定量转录组测序序列校正原理
低拷贝序列定量更准
不仅如此,还有一个重要的优势,绝对定量转录组测序其实是为低拷贝序列的准确定量而开发的。目前主流的单细胞转录组测序技术中UMI标签已被广泛应用,如大家熟悉的10x Genomics也是采用UMI标签来标记单细胞中的每个转录本并进行定量(图4)。
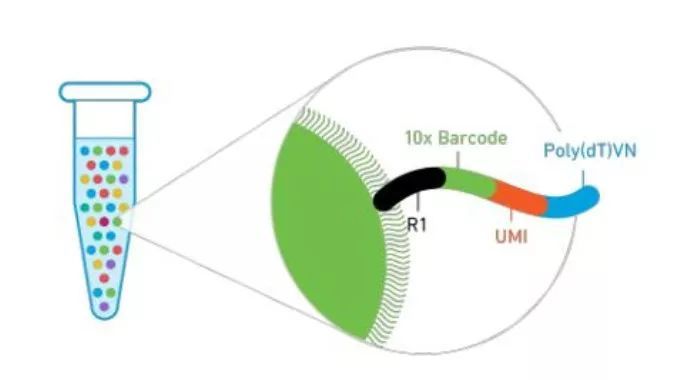
图4 10x Genomics采用UMI标记转录本
在图5中,常规转录组测序定量低拷贝序列的分布超过了3个数量级(纵轴),而绝对定量转录组测序定量的结果为1个拷贝(横轴),表明绝对定量转录组测序更胜任低拷贝序列的定量(Shiroguchi et al. 2012. PNAS)。
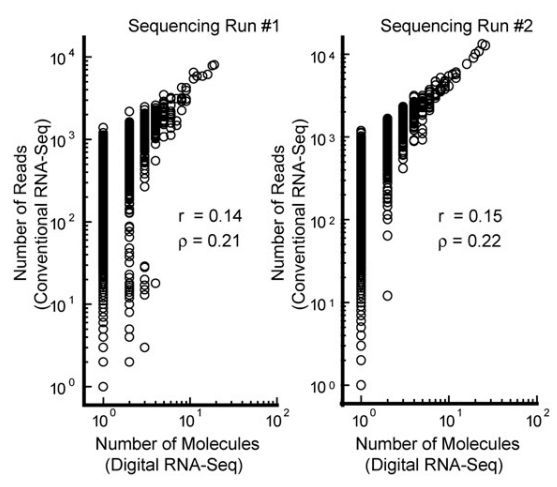
5 绝对定量转录组测序准确定量低拷贝分子
注:sequencing Run#1和#2为实验重复
好钢用在刀刃上。绝对定量转录组测序用在哪些研究中,能充分发挥她的优势呢?这就给大家介绍两个重(高)要(分)的研究领域。
互作转录组研究(需要准确的低拷贝定量)
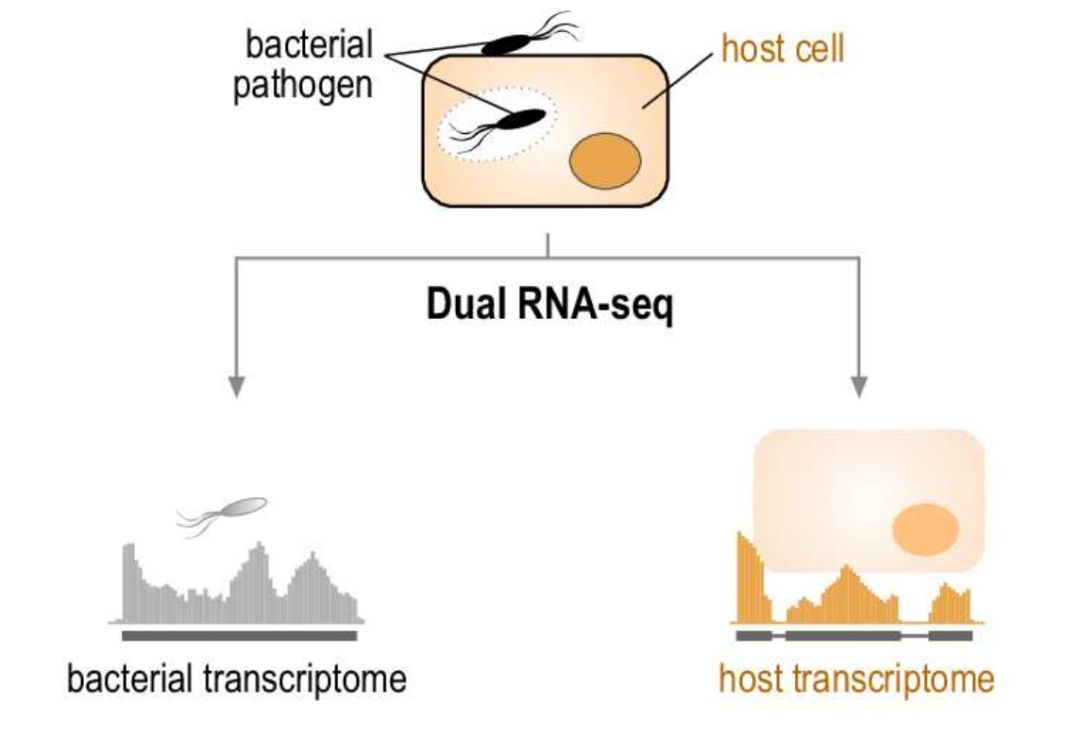
图6 Dual RNA-seq原理
自然界中存在广泛的物种间相互作用,包括寄生、共生、竞争等。在常规研究中,通常是以单一物种为主开展研究,如病菌侵染前后,研究宿主转录组的变化。但其实病菌在侵染宿主前后,它自身的转录组也会发生改变。而这种双方发生的变化对于理解病菌是如何侵染宿主,宿主又是如何应答,是非常重要的。这就是我们说的互作转录组,目前有效的检测方法是互作转录组测序(Dual RNA-seq):构建一个转录组文库即可同时对相互作用的两个物种的转录组展开研究(图6)。然而,从病菌侵染的植物叶片或动物组织中提取的RNA,绝大部分为宿主RNA,仅有极少一部分是病菌RNA。同时准确定量宿主RNA和低丰度的病菌RNA,是互作转录组研究开展的基础。以下是几篇互作转录组的研究文章,供感兴趣的小伙伴进一步了解。

本文利用Dual RNA-seq技术,研究了沙门氏菌与其感染的人类细胞中的RNA变化,揭示了一个新型小RNA(Pin-T)在调控宿主细胞转录组的分子机理。研究发现Pin-T可通过调控多种长链非编码RNA(lnRNA)和JAK-STAT信号通路,从而使得病原体更容易入侵宿主细胞且可以在宿主细胞中存活。
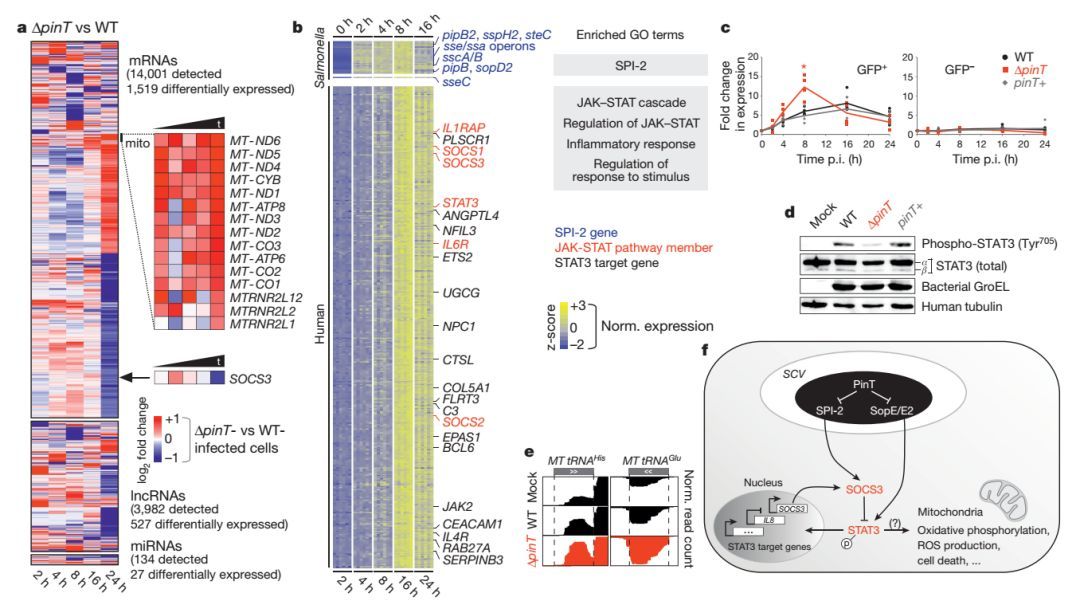
图 PinT对宿主转录组的影响

本文利用Dual RNA-seq对假结核耶尔森菌感染的淋巴组织进行研究,发现宿主转录本发生了许多变化,这些变化与炎症和急性期反应,凝血活性,以及过渡金属离子隔离有关。病原体的反应主要是为了防止吞噬攻击。耶尔森菌上调了抗吞噬III型分泌系统(T3SS)的基因表达,并诱导抵消中性粒细胞诱导的离子剥夺、自由基应激和营养抑制。
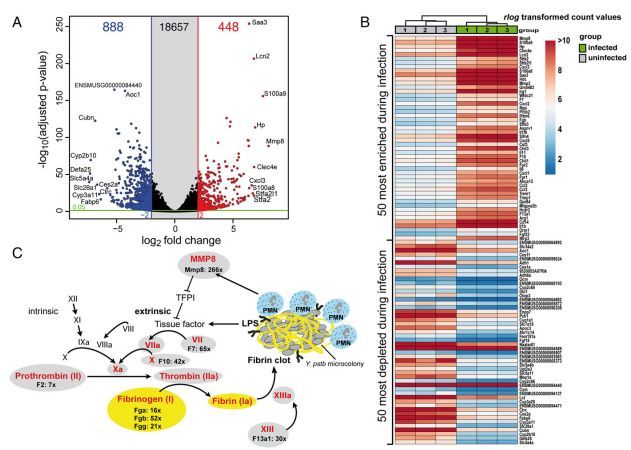
图 宿主对假结核耶尔森菌的转录水平应答

本文研究目的是鉴定在植物线虫相互作用过程中禾草根结线虫表达的致病性基因。利用Dual RNA-seq方法,测得了覆盖禾草根结线虫寄生前J2期和5个水稻寄生期的转录组学数据。在没有参考基因组的情况下,从未比对上水稻基因组的测序数据中获得禾草根结线虫转录组的66,396个contigs。对禾草根结线虫整个生命周期的基因表达谱分析,发现线虫发育的关键基因,并对寄生相关基因有了新洞察。
其他一些互作转录组研究
- Wesolowska-Andersen A, et al. Dual RNA-seq reveals viral infections in asthmatic children without respiratory illness which are associated with changes in the airway tranome. Genome Biol. (2017) 18(1):12.
- Burns AJ, et al. Tranome analysis illuminates the nature of the intracellular interaction in a vertebrate-algal symbiosis. eLife (2017) 6:e22054.
- Aprianto R, et al. Time-resolved dual RNA-seq reveals extensive rewiring of lung epithelial and pneumococcal tranomes during early infection. Genome Biology (2016) 17:198.
- Choi Y-J, et al. Dual RNA-seq of Parasite and Host Reveals Gene Expression Dynamics during Filarial Worm–Mosquito Interactions. PLoS Negl Trop Dis (2014) 8(5): e2905.
- Teixeira PJ, et al. High-resolution tran profiling of the atypical biotrophic interaction between Theobroma cacao and the fungal pathogen Moniliophthora perniciosa. Plant Cell (2014) 26(11):4245-69.
选择压力研究(需要准确的序列测定)
图7 正向遗传学与反向遗传学
从表型出发,找到与之相关的基因和突变基因型,属于正向遗传学,比如QTL定位。然而在许多研究中,因为多种因素的限制,进行精确的表型鉴定是做不到的。这时会采用另一种研究策略——反向遗传学,即直接从基因入手,找到直接影响表型性状的功能基因。
选择压力分析是反向遗传学中的重要研究方法。它的逻辑是这样的:既然所研究物种已经在不同的地域环境下发生了分化。那么,即使不知道它们精确的表型,也可以得出不同地域的个体在基因序列上会发生分化。而这些分化的基因,应该是与适应性相关的基因。
Ka/Ks比值可以判断是否有选择压力作用于蛋白质编码基因。Ka/Ks表示的是两个蛋白编码基因的非同义替换率(Ka)和同义替换率(Ks)之间的比例(不导致氨基酸改变的核苷酸变异我们称为同义突变,反之则称为非同义突变)。Ka/Ks >> 1,基因受正选择,这样的基因即为近期正在快速进化的基因,对于物种的进化有着非常重要的意义。Ka/Ks=1,基因中性进化,这样基因并没有受到自然选择压力;Ka/Ks << 1,基因受纯化选择,有害的突变通常会被淘汰。准确的蛋白质编码基因的突变鉴定,是准确的选择压力分析的基础。以下是几篇从转录组水平开展选择压力研究的文章,供感兴趣的小伙伴进一步了解。

本研究针对10种哺乳动物的6个器官进行转录组测序,覆盖了所有主要的哺乳动物谱系(胎盘类、有袋类和单孔目类)和鸟类(进化的外群),从表达水平上反映哺乳动物转录组进化的动态变化。由于受到不同的选择压力,导致基因在组织、家系,染色体水平上的进化速率不同。其中神经系统进化缓慢,睾丸组织则快速进化。啮齿动物比猿和单孔目动物进化慢,X染色体形成后快速进化。尽管哺乳动物基因表达受到强烈的纯化选择,研究发现许多潜在的选择能够驱动基因表达的变化。
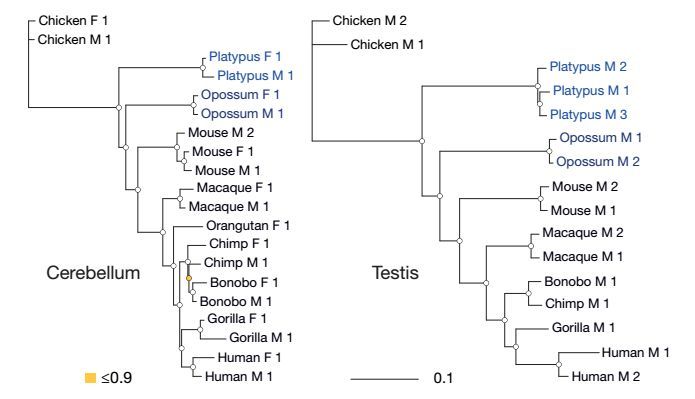
图 哺乳动物基因表达种系发生

本文对一个栽培番茄和五个野生番茄进行转录组测序,解析栽培番茄和野生番茄在基因序列和表达水平方面的变异。在转录组水平上分析了栽培番茄和野生番茄的序列差异,找到51个受正向选择的基因,这些差异基因多与环境响应和压力忍耐相关。在选择压力下,这些基因的表达水平发生很大变化,从而证实了栽培番茄和它的近缘野生番茄在转录组水平受到了人工选择和自然选择的普遍影响。
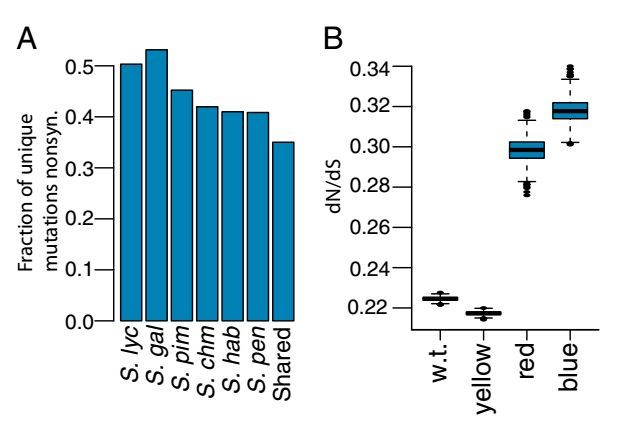
图 不同番茄品种中出现上升的非同义替代率

本文对六种墨西哥类蜀黍进行了转录组测序,发现大约75%的基因在玉米和墨西哥类蜀黍之间高度保守。发现墨西哥类蜀黍中特异表达了1516个unigenes,其中84个unigenes由4种植物的基因模型支持,571个unigenes位于玉米基因组的基因间区。鉴定出99个具有强选择信号的unigenes,57个unigenes具有>1 Ka/Ks比值,表明这些基因在玉米驯化和改良过程中可能处于强选择状态。
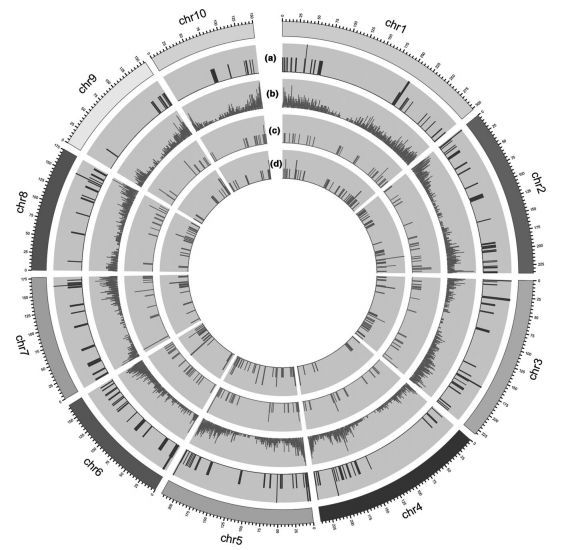
图 玉米基因组中具有不同特征和覆盖范围的unigenes分布
其他一些转录组水平选择压力研究
- Dorrell RG, et al. Progressive and Biased Divergent Evolution Underpins the Origin and Diversification of Peridinin Dinoflagellate Plastids. Mol. Biol. Evol. (2016) 34(2):361–379.
- Mao Y, et al. Comparative tranome resources of two Dysosma species (Berberidaceae) and molecular evolution of the CYP719A gene in Podophylloideae. Molecular Ecology Resources (2016) 16, 228–241
- Tao S, et al. Comparative tranome analysis and identification of candidate effectors in two related rust species (Gymnosporangium yamadae and Gymnosporangium asiaticum). BMC Genomics (2017) 18: 651.
- 本文固定链接: https://maimengkong.com/zu/1590.html
- 转载请注明: : 萌小白 2023年7月8日 于 卖萌控的博客 发表
- 百度已收录
