通过单细胞转录组测序技术得到海量细胞的表达矩阵,可以对细胞亚群做聚类、注释,鉴定亚群marker genes,寻找组间差异调控网络,绘制组织特异性图谱.....除了这些基本分析思路外,还有没有其他方法可以挖掘出隐藏在丰富数据里的宝藏呢?今天细胞轨迹分析告诉你~

一个组织样本中同时含有处于不同分化阶段的细胞,单细胞转录组测序技术一个样本可检测上千甚至上万个细胞,记录下样本中成千上万个细胞的瞬时信息,在没有多时间点样本的情况下,利用单细胞数据进行细胞轨迹分析,也能帮助构建细胞分化过程的图谱。相较传统的细胞谱系追踪方法,细胞轨迹分析可以在单细胞分辨率验证已知的细胞分化关系,简单又高效,还能推断未知的细胞分化路径,挖掘一些稀少的中间状态细胞,解析细胞分化过程中的起调控作用的关键基因,在发育生物学中细胞分化、谱系发育研究方向、肿瘤/疾病微环境中免疫细胞的动态变化研究中均有广泛应用。
如何进行细胞轨迹分析?
目前进行细胞轨迹分析的方法和软件非常之多,大致可以概括为两种方法,一种是以monocle软件为代表的 拟时序分析(pseudotime analysis),另一种则是以velocyto /scVelo为主的 RNA速度分析(RNA velocity)。
1、拟时序分析
2019年Nature biotechnology的一篇文章A comparison of single-cell trajectory inference methods [1] 就对45种拟时序分析的方法进行了评估,这些软件在性能、可扩展性、稳定性和易用性上存在差异,当然也各有其优缺点。软件推断的细胞分化轨迹主要有7种类型,包括环状(cycle)、线性(linear)、分叉(bifurcation)、多分叉(Multifurcation)、树型(Tree)、多种轨迹并存的连接图(Connected graph),及多种不相连轨迹并存的断开图(disconnected graph)。(图1)
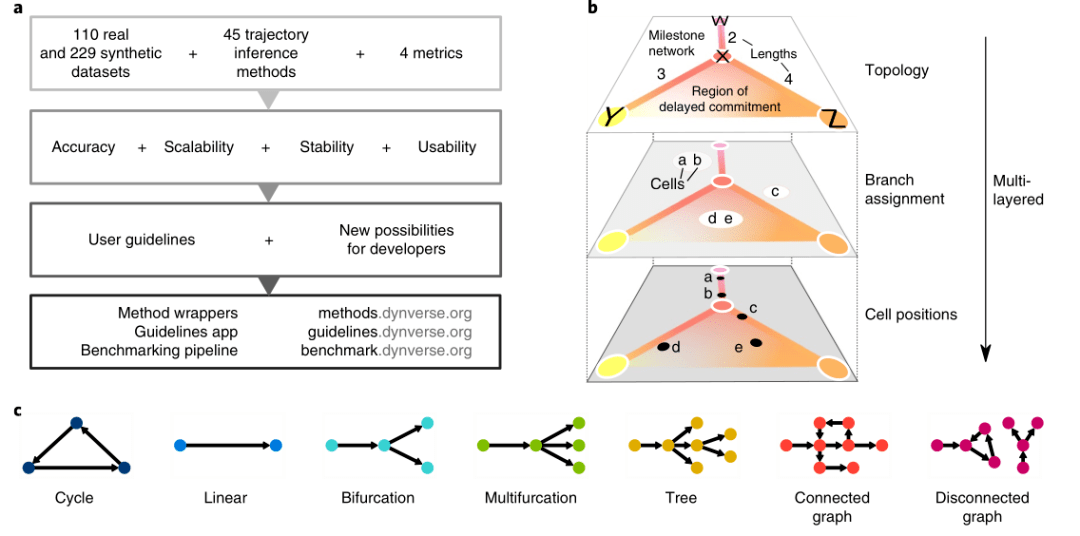
图1 轨迹分析算法评估流程及7种细胞分化轨迹类型[1]
Monocle2 [2]是目前引用率比较高的一款软件,基于基因表达量的动态变化来构建细胞轨迹,软件简单易操作,且功能较全面,能满足基本分析所需的细胞分化轨迹类型;软件更新速度也比较快,如果细胞通量较高会严重影响Monocle2的运行速度,进行大数据集分析时需要选择特定细胞类型来减少计算量,这个问题在Monocle3中得到了解决。Monocle2会默认分化轨迹的起点,但这个起点不一定符合生物学意义,所以需要基于样本自身的生物学背景,挑选已知或潜在存在分化关系的细胞类型显得尤为重要,来明确正确的细胞分化起点和终点。
2、RNA速度分析
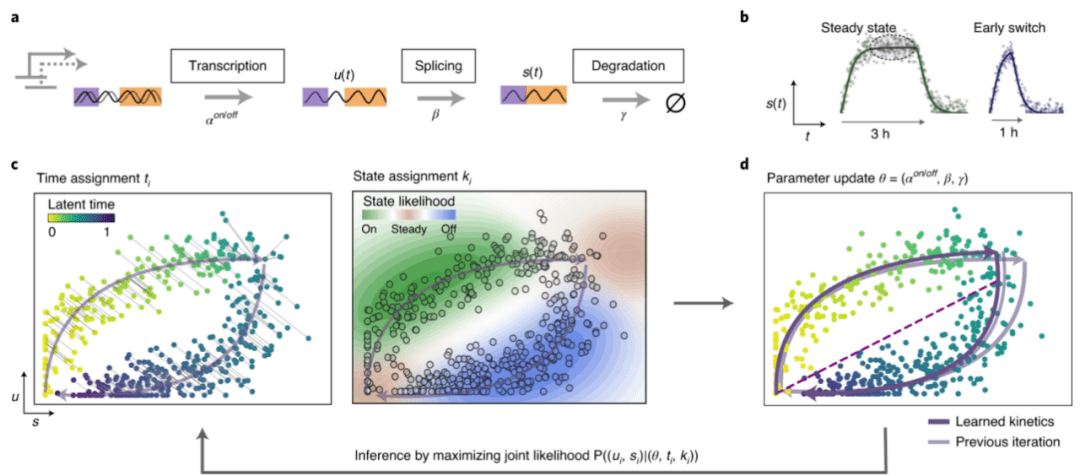
图2 scVelo软件分析原理示意图[4]
a.转录动力学建模捕捉未剪接前 mRNA 的转录激活和抑制(“开”和“关”阶段)这些转录本转化为成熟的mRNA并最终降解;b.当转录激活和抑制的转录阶段分别持续足够长的时间时,分别达到活跃转录和非活跃稳态。然而,特别是在瞬态细胞群中,通常不会达到稳定状态,例如,转录激活可能在 mRNA 水平饱和之前终止,表现出"提早转换”行为;c. 基于似然的模型,解决剪接动力学的完整基因转录动力学,它由两组参数控制:(1)转录、剪接和降解的反应速率(2)细胞-转录状态和时间的特定潜在变量。通过 EM 迭代地推断参数。对于给定的反应速率参数估计,通过最小化每个单元与当前相轨迹的距离来将时间点分配给每个单元。转录状态是通过将可能性与轨迹上的各个片段相关联来分配的——即激活、抑制以及活跃和不活跃的稳态。d.然后通过更新反应速率的模型参数来优化整体可能性。紫色虚线将推断的(未观察到的)非活动状态与活动稳定状态联系起来。
相较于拟时序分析来说,RNA velocity分析的是细胞在某一时刻的潜在分化方向,可以直接从具有时间向量的结果读取到分化方向,无需基于生物学背景指定分化起点和终点;同时RNA velocity的数据可以与Seurat降维数据兼容,也可以嵌入monocle构建细胞轨迹,这对于联合两种分析策略十分便利,利用RNA velocity分析揭示所有未知的细胞分化关系,然后筛选关注的细胞分化过程进行拟时序分析,挖掘分化过程中的调控机制。
百迈客可提供以上两种细胞轨迹分析,有需要的老师可联系百迈客当地销售,或在评论区留言, 欢迎各位老师前来咨询。
如何应用细胞轨迹分析 ?
上文有提过,细胞轨迹分析目前在细胞分化、谱系发育、肿瘤/疾病微环境中免疫细胞的动态变化研究中均有广泛应用,大致可以概括为以下三种应用场景:
1、细胞分化过程重现
基于单细胞测序分析数据,对其他技术手段获取的已知细胞分化关系进行验证。
2020年Nature Communication发表的Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy[5],利用RNA速度表征了胶质母细胞瘤的分化过程,证实了胶质母细胞瘤层次结构顶点是肿瘤祖细胞,星形细胞、间充质、少突胶质细胞和神经元癌细胞比肿瘤祖细胞的分化程度更高,并且在每个患者中均可见特定细胞分化方向(图3)。
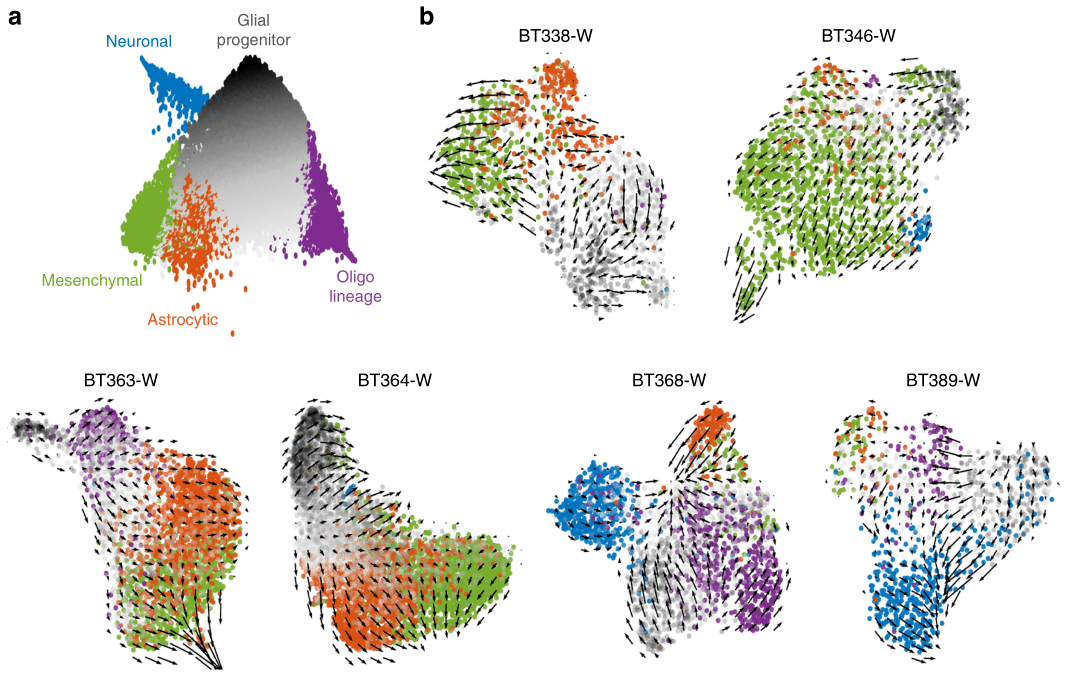
图 3 胶质母细胞瘤RNA速度分析[5]
2020年Cell stem cell发表的The Dynamic Tranional Cell Atlas of Testis Development during Human Puberty[6],研究根据前期研究基础将人睾丸生殖细胞分为对应4个发育阶段的细胞亚型:未分化的精原细胞、分化的精原细胞、精母细胞、精子细胞;同时利用拟时序分析,证实了生殖细胞由未分化的精原细胞向精子细胞分化的发育轨迹(图4)。
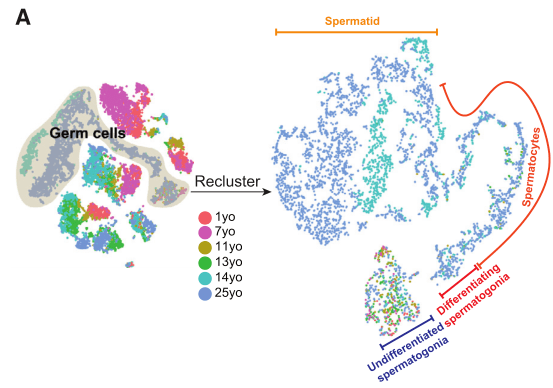
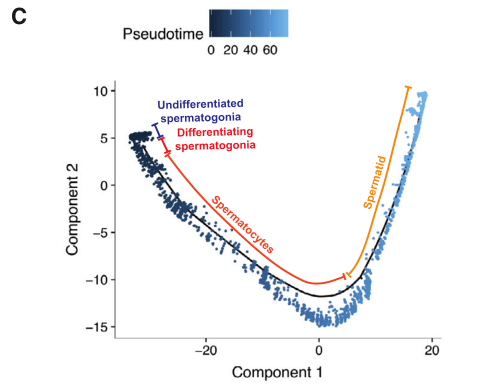
图4 人睾丸生殖细胞亚群及分化轨迹鉴定[6]
2、潜在的细胞分化路径挖掘
除了鉴定已知的细胞分化过程,通过细胞轨迹分析也能推断未被报道的、可能的未知细胞分化路径,如神经干细胞发育、肿瘤/疾病微环境中免疫细胞分化过程,甚至挖掘到一些稀少的、常规技术手段很难获得的中间状态细胞。
2021年Nature Communication发表的Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis[7],研究首先通过比较两种亚型类风湿性关节炎(RA)患者与健康对照外周血免疫细胞图谱差异,关注到了B细胞类型丰度变化,发现ACPA-RA组IGHG4+ plasma B细胞明显升高,HLA-DRB5+ plasma B细胞明显降低(图5,左图);随后对B细胞进行拟时序分析探究B细胞亚型分化关系,发现在健康对照和RA患者之间B细胞亚型存在不同的分化路径:在健康对照中naïve B细胞有两条分化路径,1条是分化为HLA-DRB5+plasma B细胞再分化为plasma B细胞,另1条分化为Mem-unsw B细胞到Mem-sw B细胞;而RA患者中,naïve B细胞直接分化为IGHG4+ plasma B细胞(图5,右图)。
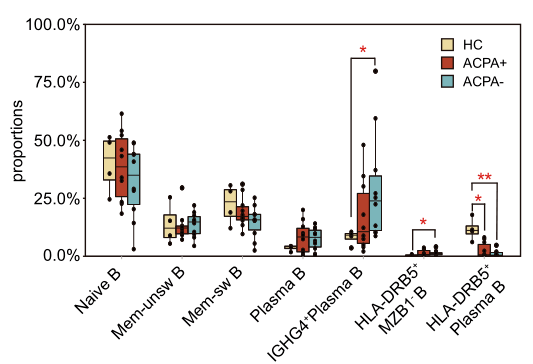
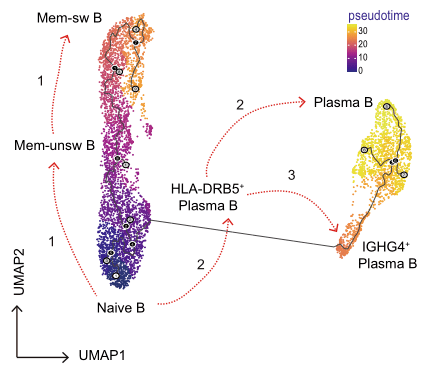
图5 类风湿关节炎患者外周血B细胞分析[7]
胶质母细胞瘤 (GBM) 可以根据基因表达分为不同的亚型,但尚未鉴定出经典亚型的胶质瘤干细胞样细胞 (GSC),2019年Cancer Discovery发表的The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation[8],对IDH 野生型 GBM单细胞数据进行 RNA 速度分析,鉴定到了GBM的肿瘤干细胞样细胞 (GSC)群体逐渐从间充质表型mGSC 过渡到原神经表型pGSC的中间群体,在这个中间群体中,mGSC 标记(如CD44、CHI3L1)高表达;而pGSC 标记物(如DLL3、PDGFRA)的表达较低,但具有较高的正表达速度(图6)。
图6 胶质母细胞瘤干细胞样细胞分化轨迹[8]
3、细胞分化过程调控机制解析
通过细胞轨迹分析,分析处于重要分化节点的关键细胞亚群,寻找随着分化时间呈现特定表达模式的关键基因,解析驱动细胞亚群分化的调控机制。
2021年 Nature发表的Single-cell tranomic characterization of a gastrulating human embryo[9],为了探索原肠胚形成过程中外胚层的变化,利用RNA速度分析了外胚层(Epiblast)、外胚层(羊膜/胚胎)(Ectoderm (amniotic/embryonic))、新生中胚层(Nascent mesoderm)、原条(Primitive Streak),由Epiblast起,发生两个分支,一侧由Primitive Streak发育到Nascent mesoderm,一侧发育到ectoderm(图7,左图);随后利用拟时分析推断Epiblast分化过程中的基因表达变化,检测到ectoderm marker genes(DLX5、TFAP2A 和GATA3)表达上调,而早期神经诱导marker genes(SOX1、SOX3 和PAX6)和分化的神经元marker genes(TUBB3、OLIG2和NEUROD1)表达极低甚至检测不到,这表明CS7中神经分化尚未开始(图7,右图)。
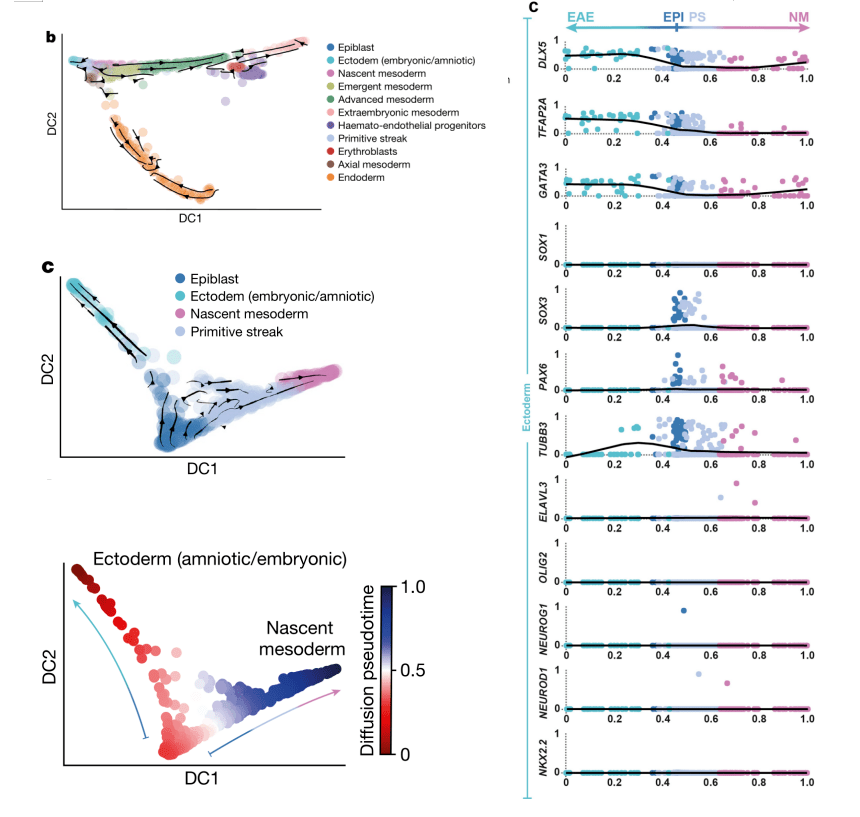
图7 RNA速度分析解析原肠胚分化轨迹[9]
如何进行RNA速度分析 ?
1、软件安装:
pip install-U scvelo #需要3.6以上版本python
pip installvelocyto
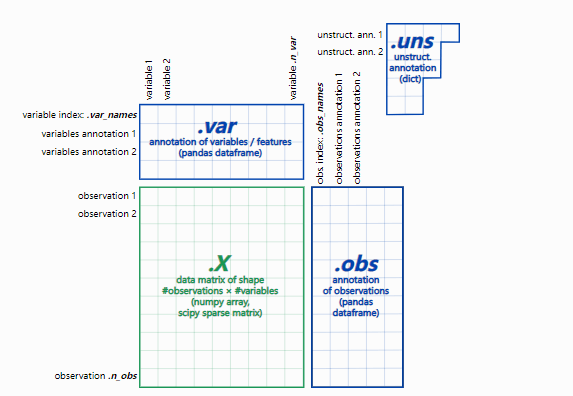
2、adata 数据结构:
– adata.X:数据矩阵
– adata.obs:细胞的注释
– adata. var:基因的注释
– adata.nus:非结构化注释,例如图
– adata.layers:附加层数据,如spliced/unspliced矩阵
3、加载需要的库及一些配置
importseaborn assns
importscvelo asscv
importpandas aspd
importscanpy assc
importnumpy asnp
importloompy
scv.settings.verbosity = 3# show errors(0), warnings(1), info(2), hints(3)
scv.settings.presenter_view = True# set max width size for presenter view
scv.set_figure_params( 'scvelo') # for beautified visualization
4、计算获取unspliced/spliced矩阵
## 运行run10x
/software/python3.9.6/bin/velocyto run10x \
# 相应的repeatmasker的重复序列注释文件(可从USCU中获取)
# http://genome.ucsc.edu/cgi-bin/hgTables
-m RMSK
cellranger_results # cellranger的输出结果路径
sample_name # 样本名
seurat_GTF # 用于cellranger定量的基因组注释文件
## 输出结果,在cellranger的结果中会创建单个样本的velocyto文件夹,其中的loom文件即为最终结果
data/SC/Normal1-sc/velocyto:
Normal1-sc.loom
data/SC/Normal2-sc/velocyto:
Normal2-sc.loom
data/SC/Normal3-sc/velocyto:
Normal3-sc.loom
data/SC/Normal4-sc/velocyto:
Normal4-sc.loom
5 、loom文件的读取及合并
# 读取单个样本loom文件
Normal_loom = sc.read('./Normal1.loom', sparse=True, cache= True)
# 合并同组样本的loom文件
importloompy
Normal_loom_list = [Normal1-sc.loom,
Normal2-sc.loom,
Normal3-sc.loom,
Normal4-sc.loom]
loompy.combine(Normal_loom_list, output_file = 'Normal_combined.loom')
# 读取
Normal_loom = sc.read( './Normal_combined.loom', sparse= True, cache= True)
6 导入Seurat对象的降维聚类结果
使用 scVelo进行分析时需要对数据进行 降维聚类,若上游分析的降维聚类注释结果基于 Seurat,可以直接将 Seurat的降维聚类结果添加到 adata对象中,下面方法仅供参考。此外,如果上游分析基于 scanpy,则可以直接调用 scanpy的降维聚类相关的方法。 注:此处并没有进行注释,在实际操作中将 seurat_cluster替换为注释后的信息即可,导入seurat之后的所有调用的方法中的groupby参数设置为 seurat_cluster。
library(Seurat)
library(tidyverse)
seurat_obj@meta.data %>% rownames_to_column(var = 'barcodes') %>%
write.table( 'seurat_meta_df.txt', sep = '\t', quote = F)
seurat_obj@reductions$umap@cell.embeddings %>% rownames_to_column(var = 'barcodes') %>%
write.table( 'seurat_umap.txt', sep = '\t', quote = F)
defusing_seurat_meta_umap(loom_file, seurat_meta_df, seurat_umap):
# 读入seurat的meta.data以及umap降维结果
seurat_meta_data_raw = pd.read_csv(seurat_meta_df)
seurat_umap_raw = pd.read_csv(seurat_umap)
# loom文件原始的index顺序
anndata_index = loom_file.obs.index.values
# 合并
meta_data = loom_file.obs.reset_index. \
merge(seurat_meta_data_raw,
left_on= "CellID",
right_on= "barcodes", how= 'left'). \
set_index( 'CellID').reindex(anndata_index)
meta_data[ 'seurat_clusters'] = meta_data[ 'seurat_clusters']. \
apply(lambda x:"%d"% int(x) if~np.isnan(x) elsex).astype( 'category')
loom_file.obs = meta_data
loom_file = loom_file[loom_file.obs.dropna.index, :]
loom_file.obsm[ 'umap'] = seurat_umap_raw.set_index( 'barcodes'). \
reindex(loom_file.obs.index).to_numpy.astype( 'float64')
returnloom_file
Normal_loom = using_seurat_meta_umap(Normal_loom,
'seurat_meta_df.txt',
'seurat_umap.txt')
# 查看是否替换成功,与seurat结果进行比较
sc.pl.umap(Normal_loom, color= 'seurat_clusters')
分析结果展示
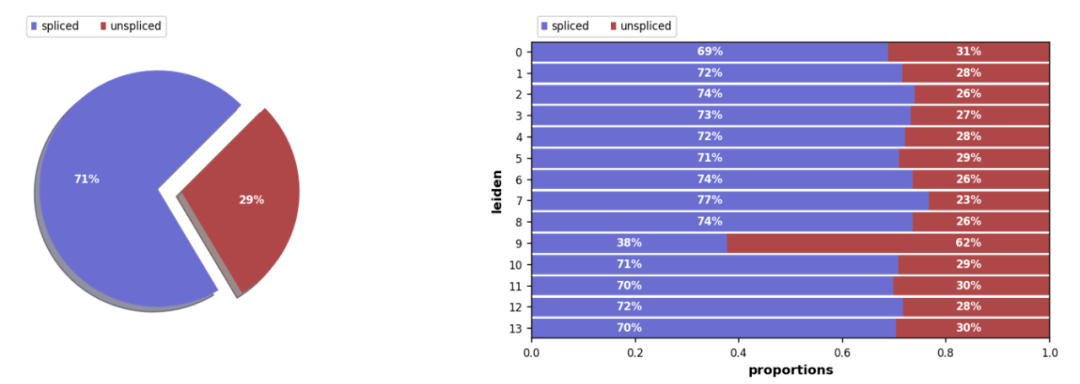
拼接/未拼接计数的比例,通常有 10%-25% 的未拼接分子包含内含子序列。
scv.pl.proportions(Normal_loom, groupby= 'seurat_clusters')
RNA-速度:速度是基因表达空间中的向量,代表单个细胞运动的方向和速度。速度是通过模拟剪接动力学的转录动力学获得的,对于每个基因,拟合了成熟(未剪接)和成熟(剪接) mRNA计数的稳态比率,这构成了恒定的转录状态。正速度表明基因被上调,这种情况发生在该基因未剪接 mRNA丰度高于预期的稳定状态的细胞中。相反,负速度表明基因被下调。
# 预处理
scv.pp.filter_genes(Normal_loom, min_shared_counts= 20)
scv.pp.normalize_per_cell(Normal_loom)
scv.pp.filter_genes_dispersion(Normal_loom, n_top_genes= 2000)
scv.pp.log1p(Normal_loom)
scv.pp.filter_and_normalize(Normal_loom,
min_shared_counts= 20,
n_top_genes= 2000)
# 运行scVelo动态模型,如果算力不够可以考虑静态或者随机方法
scv.tl.recover_dynamics(Normal_loom, n_jobs= 30)
scv.tl.velocity(Normal_loom, mode= 'dynamical')
scv.tl.velocity_graph(Normal_loom, n_jobs=n_jobs)
# 保存计算结果
Normal_loom.write( 'Normal_dy_model.h5ad', compression= 'gzip')
scv.pl.velocity_embedding_stream(Normal_loom,
basis= 'umap',
color= 'seurat_clusters')
scv.pl.velocity_embedding(Normal_loom,
arrow_length= 3,
arrow_size= 2,
show= False,
color= 'seurat_clusters')
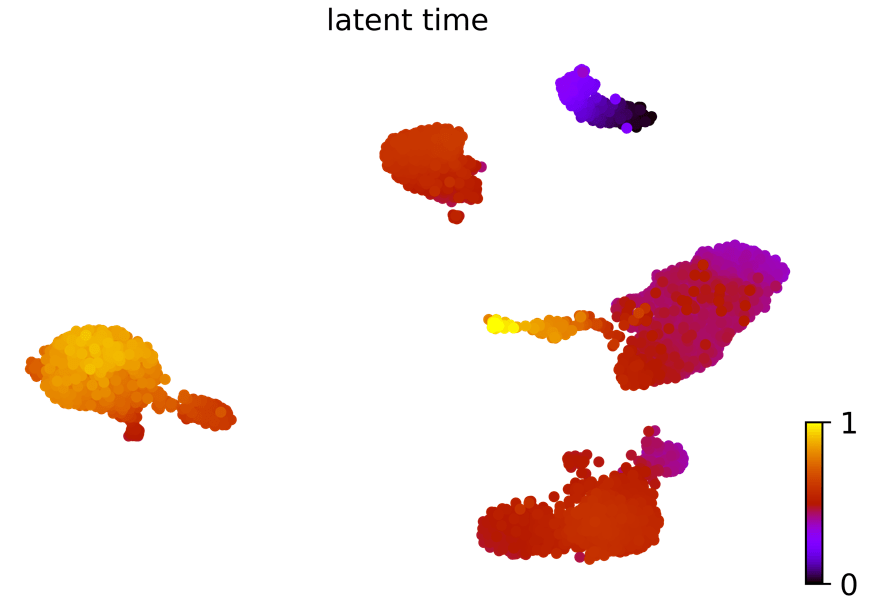
潜伏时间:动力学模型描述了细胞过程的潜伏时间(这个潜伏时间代表细胞的内部时钟,并近似于细胞在分化时经历的真实时间,仅基于其转录动力学。)
scv.tl.latent_time(Normal_loom)
scv.pl.scatter(Normal_loom, color= 'latent_time',
color_map= 'gnuplot', size= 80, show= False)
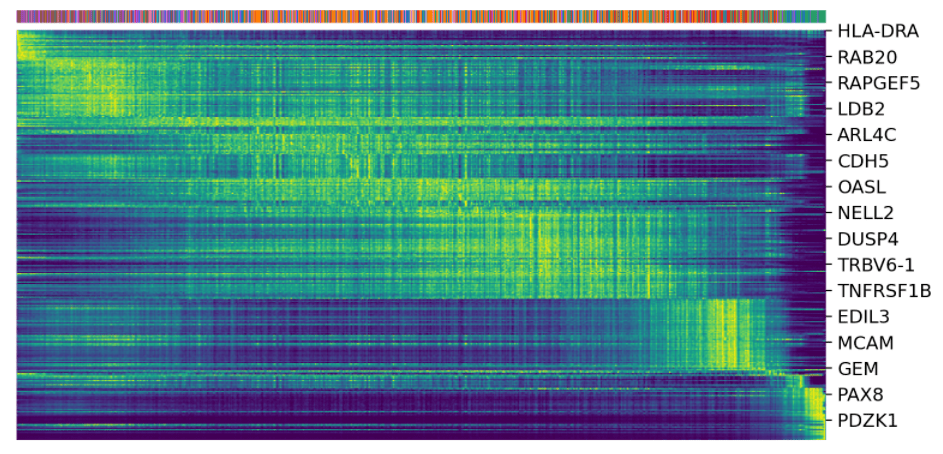
驱动基因:显示出明显的动态行为,并通过动态模型中的高可能性特征系统地检测到。
top_genes = Normal_loom. var[ 'fit_likelihood']. \
sort_values(ascending= False).index[: 300]
scv.pl.heatmap(Normal_loom,
var_names=top_genes, sortby= 'latent_time',
col_color= 'seurat_clusters', n_convolve= 100)
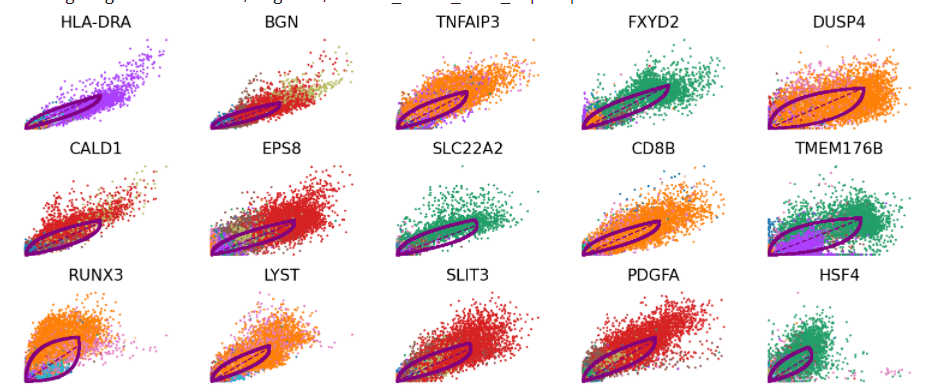
scv.pl.scatter(Normal_loom, basis=top_genes[: 15],
ncols= 5, frameon= False,
color= 'seurat_clusters')
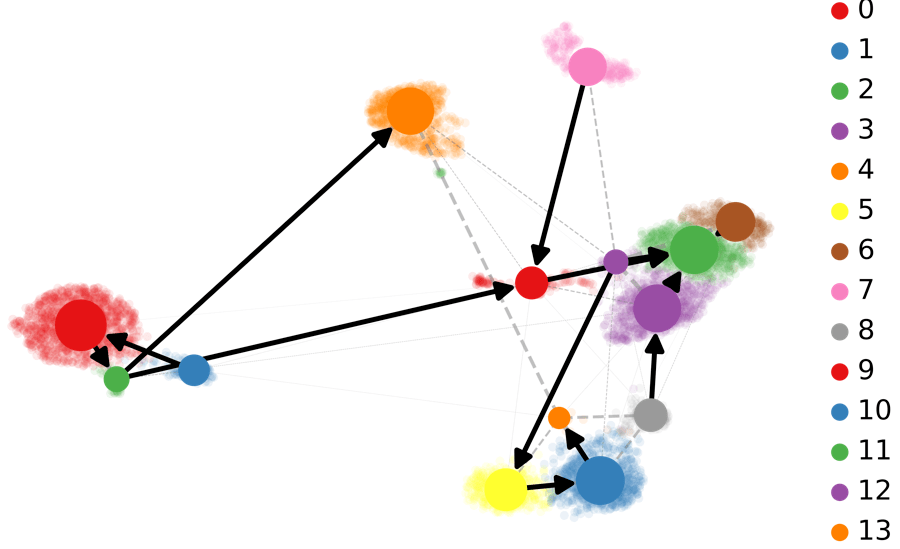
基于RNA-速度的PAGA分析:PAGA提供了轨迹推断构图的方法,其中加权边对应于两个集群之间的连接。在这里,PAGA通过速度推断的方向性进行了扩展。
scv.tl.paga(Normal_loom, groups= 'seurat_clusters')
scv.pl.paga(Normal_loom, basis= 'umap',
size=50, alpha=.1,
min_edge_width=2, node_size_scale=1.5)
END
百迈客是国内首批引进10x genomics单细胞测序平台,在国内展开高通量单细胞测序服务。目前累计成功制备200余种组织类型、5000+例的样本,覆盖的物种和样品类型包括人和小鼠的脑组织、肺脏、皮肤、视网膜、肾脏、脾脏、小肠,以及斑马鱼、拟南芥、水稻等。为了帮助更多老师开展单细胞空间多组学研究领域,资深单细胞技术团队在百迈客医学微信公众号创办了 百迈客单细胞 & 空间转录组专题栏目系列,可提供专业的课题方案设计、案例分享、数据分析和时空多维解决方案,同时含有 发文思路、分析技巧、项目经验等应有尽有 !
如果你喜欢本文欢迎点赞收藏转发,你的鼓励是我们分享的动力,谢谢!祝您科研顺利!
参 考
[1] Saelens, Wouter et al. “A comparison of single-cell trajectory inference methods.” Nature biotechnology vol. 37,5 (2019): 547-554. doi:10.1038/s41587-019-0071-9
[2]http://cole-trapnell-lab.github.io/monocle-release/docs/
[3]La Manno, Gioele et al. “RNA velocity of single cells.” Nature vol. 560,7719 (2018): 494-498. doi:10.1038/s41586-018-0414-6
[4]https://scvelo.readthedocs.io/
[5]Couturier, Charles P et al. “Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy.” Nature communications vol. 11,1 3406. 8 Jul. 2020, doi:10.1038/s41467-020-17186-5
[6]Guo, Jingtao et al. “The Dynamic Tranional Cell Atlas of Testis Development during Human Puberty.” Cell stem cell vol. 26,2 (2020): 262-276.e4. doi:10.1016/j.stem.2019.12.005
[7]Wu, Xunyao et al. “Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis.” Nature communications vol. 12,1 4977. 17 Aug. 2021, doi:10.1038/s41467-021-25246-7
[8]Wang, Lin et al. “The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation.” Cancer discovery vol. 9,12 (2019): 1708-1719. doi:10.1158/2159-8290.CD-19-0329
[9]Tyser, Richard C V et al. “Single-cell tranomic characterization of a gastrulating human embryo.” Nature vol. 600,7888 (2021): 285-289. doi:10.1038/s41586-021-04158-y
转自:百迈克
- 本文固定链接: https://maimengkong.com/zu/1535.html
- 转载请注明: : 萌小白 2023年5月13日 于 卖萌控的博客 发表
- 百度已收录
