10X genomics ScRNA-seq数据的分析,属于群体单细胞数据分析的范畴。分析大体可以分为以下四个步骤(图1)。除了数据质控与过滤属于二代测序通用的步骤外,后续的三个步骤是群体单细胞转录组分析特有的内容,我们下文来一一解析。
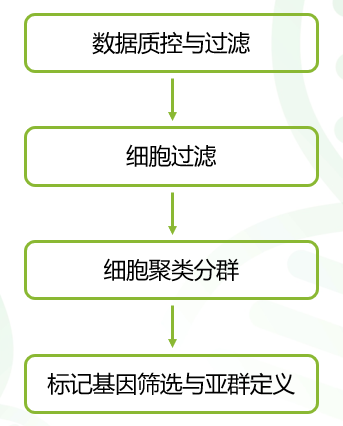
图1 基础分析步骤
细胞过滤
10X ScRNA-seq数据可以每个样本获得数千个单细胞的转录组信息。但并非每个单细胞的数据都是合格的,因此需要设立标准进行过滤。需要过滤掉的细胞数据包括:
1
来源空载(油包水磁珠(GEM)数据
10X 单细胞数据来源一个油包水磁珠(GEM)为单元的单细胞扩增。约有65%的GEM在10X芯片的管道中可以成功捕获细胞,但也有35%没有捕获细胞。当然,空载的GEM并非没有任何结果。由于单细胞悬液中存在细胞破裂产生的游离RNA,因此空载GEM中也会扩增得到少量RNA的信息,但基因数会很少。
2
过载的GEM数据
部分GEM在10X芯片中可能也会出现过载,既1个GEM中吸入了两个或两个以上的细胞。这样两三个细胞信息就被合并了,从而相互干扰。
3
状态异常的细胞信息
这部分常常是实验操作过程中衰亡自噬的细胞,这类细胞已经不具有研究的价值,应该被去除。
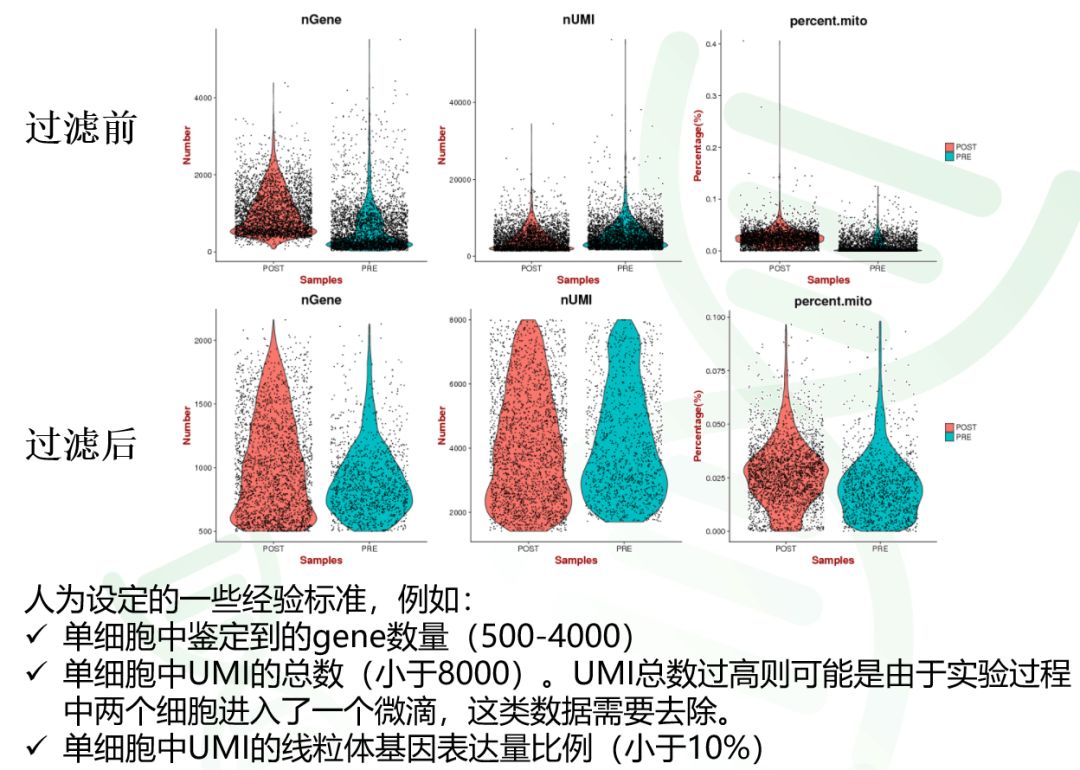
图2 细胞过滤前后的比较
对于以上问题,我们基迪奥生物主要通过以下三个指标进行过滤,从而去除不合格的单细胞数据(合格比单个追求细胞数量更重要)。
细胞中检测到的基因数,太低或太高的细胞将被过滤
检测基因数太少的单细胞数据,可能来自空载的GEM。检测基因数太高的,则可能来自过载的GEM。
细胞中检测到的UMI总数,太低或太高的细胞将被过滤
UMI就是cDNA扩增前给每个cDNA连上的一个标签序列。所以一段UMI序列,就代表一分子的cDNA。1个细胞UMI的总数就代表细胞中初始mRNA的总量。同样的,过滤mRNA总量太低或太高的细胞,可以有效去除空载或过载的GEM。
线粒体基因表达量比例过高的细胞被去除
自噬凋亡的细胞,通常会出现线粒体基因的超表达。因此,利用这一指标,可以去除此类细胞。
细胞聚类分群
1
亚群分类的方法
细胞亚群分类是10X ScRNA-seq数据分析的核心步骤,不同软件有不同的算法。因为10X ScRNA-seq数据量大,数据结构复杂,对应的分群算法也比较复杂,我们这里就不讲具体的算法,只是简单说下最基本的原理。
分群的基本原理就是利用基因表达量的信息,计算各个细胞间表达模式的差异度,然后基于一定的标准将所有细胞归为多个亚群(将差异度小于值的细胞归为一个亚群)。
要注意的是,这个阶段的细胞分群还只是一个存粹数学的结果,会随着参数的调整而不断变化。所以,每个参数分析的结果,并不一定就符合生物预期。分群结果是否符合预期,还需要后续基于特征标记对各个亚群进行定义后才可以判断。
细胞分群结果一般要进行可视化展示。说到单细胞结果可视化,就不得不提起tSNE映射图。
2
亚群分类映射图以及tSNE与PCA的比较
亚群分类结果展示,经常采用散点映射图的方式。这个过程可以分为分两步:
(1)先用某种算法(例如PCA或tSNE)实现将细胞间的关系用散点图进行展示(图3.ab);
(2)根据某些指标(例如上一步获得的细胞分群结果)将散点图中的点(代表细胞)涂上不同颜色,这个步骤称为映射(Project)。例如,图3.cd就是对属于11个亚群的点分别涂上11种颜色。
那么,接下来我们来讨论下tSNE算法与PCA的区别。
先说结论:PCA是常规转录组常用的数据降维和样本关系可视化的方法。但针对群体单细胞转录组数据,tSNE是明显胜过PCA的方法。
tSNE也是一种数据降维的方法,方法上非常类似PCA(主成分分析)。关于PCA分析的介绍可以参考我们Omichare论坛的介绍:
http://www.omicshare.com/forum/forum.php?mod=viewthread&tid=631&highlight=PCA
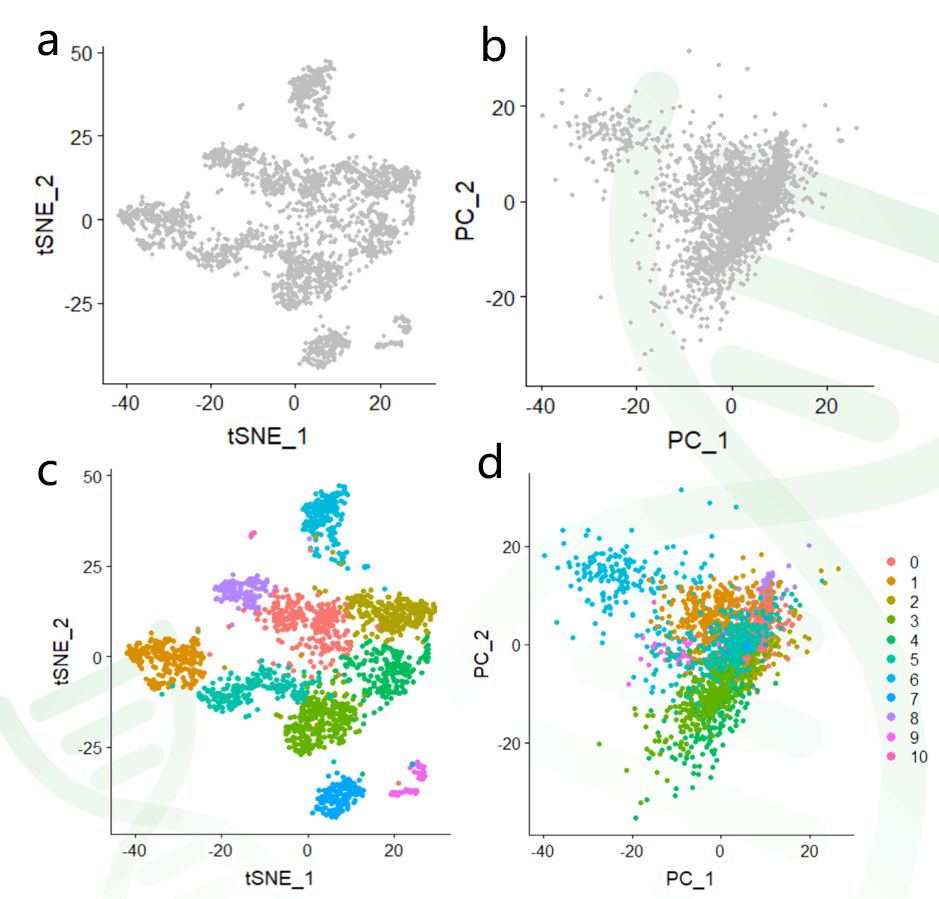
图3 利用tSNE(a)以及PCA(b)的方法对单细胞群体数据降维度并绘图,然后对不同细胞亚群的点涂上不同的颜色(c,d)
那么针对转录组数据,tSNE和PCA有什么区别呢?转录组数据是一种非常复杂的数据集,其检测基因数大。而且,随着检测样本数的增加(例如,10XScRNA-seq一次检测成千上万个细胞),数据复杂程度会大大提高。
PCA是线性降维的方法。以转录组数据为例,PCAd的主要目的是从大量基因的表达量信息中,提取对整体基因表达量影响最大的效应(例如,实验处理效应,实验批次效应,异常样本效应等)。这些效应被称为主成分(PC),然后利用主成分绘图(图3b)。
简而言之,PCA分析就是将数据中大量基因的信息浓缩到少数几个变量(代表样本中的主要效应)中。而后,只要2~3个变量(命名为PC1,PC2,PC3)就可以代表原来几万个基因含有的大部分信息。那么细胞之间表达量差异,就体现在PC1、PC2这些变量数值上的差异。
最后,我们只要用PC1和PC2绘制散点图,就可以将有差异的细胞区分开。如图3b,每个小点代表一个细胞,利用每个细胞PC1和PC2值绘图。
为什么PCA适用于常规RNA-seq而不大适用于10X ScRNA-seq呢?
PCA的方法侧重于去抓住样本中隐含的主要效应,从而让差异大的样本(即,在主要效应上差异大的样本),在图中呈现较远的距离。在常规RNA-seq项目中,一般样本不多,实验处理效应组合数通常不会超过10种(例如,2类病人× 3个时间点取样 = 6种处理组合),因此每个实验处理效应在所有因素的总体效应中占比都比较大,属于效应比较大的因素。
另外,实验批次效应,离群样本等也属于比较大的效应。以上的效应都易于被PCA获取,因此 PCA的方法可以良好地去展示常规RNA-seq项目中的处理效应、批次效应和离群样本。
但是如果影响样本分组的不是主要效应,而是一些效应更小的效应,PCA则无法对这些样本进行准确区分。而10X ScRNA-seq的可视化展示,主要期望对各个细胞亚群有良好的区分。在每次检测的上万个细胞中,几乎肯定可以区分出几十种细胞亚群,包括一些稀有的细胞。
区分这些细胞亚群(尤其是稀有细胞类型)的效应,往往不是主要效应(即大量基因的差异),而是一些微小效应(少量标记基因差异)。那么,PCA的方法就无法将这些细胞亚群良好区分。从图3.bd中我们也可以看到PCA对细胞亚群有一定的区分能力。但分类结果有缺点:属于一个亚群的细胞比较离散,因此不同亚群间存在明显重叠,总体展示效果不佳。
那么,什么样的算法最理想的算法呢?最理想的算法,应该是能够兼顾两点:
1)局部结构:属于同一个亚群的细胞,聚类尽可能近
2)全局结构:属于不同亚群的细胞,聚类尽可能分开
tSNE算法就属于这种可以同时兼顾局部结构和全局结构的非线性降维可视化算法。从图3.ac的结果中我们可以看到,在tSNE绘图结果中,属于同一个亚群的细胞紧密成簇,而属于不同亚群的细胞则有明显边界。
总而言之,tSNE不同于PCA(PCA主要目标是尽量去抓取群体中的主要效应),tSNE算法的主要目标是:在经历数据降维后(例如从2万基因降低到2个变量),在原始2万个基因的数据集中最相似的细胞,在降维后的数据中依然保持最为相似(紧密成簇)。这就保证了哪怕某些稀有细胞只有少量基因区别于其他细胞亚群,在tSNE中依然可以与其他细胞有良好的区分。
3
其他类型的tSNE映射图
除了亚群分组的信息,其他细胞信息也可以通过颜色的方式映射到tSNE图中,展示不同的生物学问题。
例如,细胞样本的来源。图4.a就以颜色标记了来源白血病患者骨髓移植前(红色)和移植后(绿色)样本的细胞,从而我们可以直观了解不同样本来源的细胞的亚群分布有哪些特性。
另一种常见的映射方式就是以某个标记基因的表达量给各个点上色,从而可以直观观察该标记基因主要在哪些细胞亚群中表达。如图4b,就展示了标记基因GATA1在细胞群体中的表达分布。
下一期文章,我们就要谈谈群体单细胞RNA-seq的标记基因筛选。
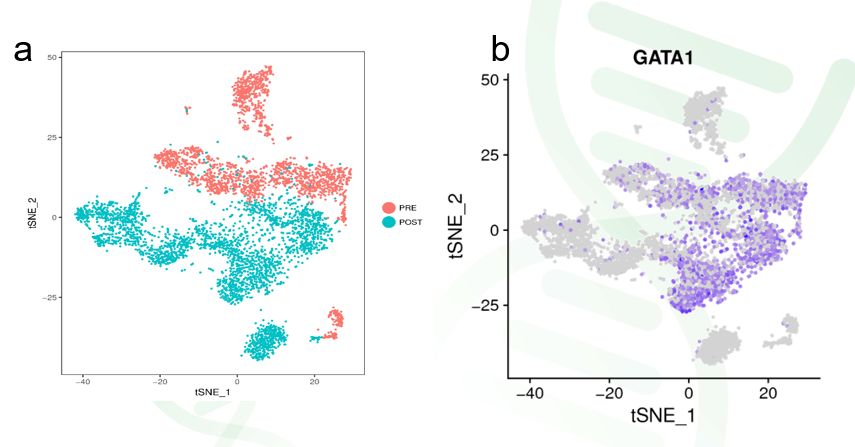
图4 基于其他信息的单细胞数据tSNE映射图
- 本文固定链接: https://www.maimengkong.com/kyjc/725.html
- 转载请注明: : 萌小白 2021年7月28日 于 卖萌控的博客 发表
- 百度已收录
