跟着Nature Communication学绘图
之Figure 1A + Supplementary Figure 1A
大家好,我是阿琛。在上一期内容中,我们介绍了如何下载TCGA数据库,TARGET数据库,CCLE数据库等来源的表达数据,以及注释信息的初步整理(“ 首发!跟着IF12的NC学单细胞测序分析,超详细!专为小白定制!(附代码) ”)。

一图胜千言。数据下载完成后,接下来我们先需要对其进行进一步的处理与分析,最终整理成为可用于绘图的格式,并将其以图的形式呈现给大家欣赏。
下面,我们一起来看下如何进行Seurat对象的构建和可视化展示。
一、R包读取
library(magrittr) library(tidyverse) library(Seurat) library(ggplot2)
正式开始之前,根据数据清洗,构建Seurat对象,以及绘图的需要,我们分别加载了tidyverse包,Seurat包,以及ggplot2包。
source( "global_params.R") source( "analysis_helpers.R")
同时,作者提供了几个相关的自定义函数,用于后续的分析使用。
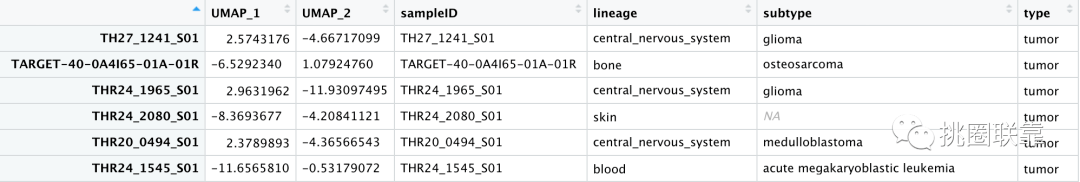
结果显示:
二、读取数据
load( "result_NC.Rdata")
接着,我们将肿瘤和细胞系的表达数据及其注释文件读取进来。关于这部分数据的下载与整理,在上一期的内容中已经进行了相关的介绍,并将最后的结果保存为.Rdata文件。因此,直接使用load函数即可将结果读取进来。
结果显示,其中包含了结果文件result和注释信息文件hgnc.complete.set。

点击环境变量Environment中的result,或者使用代码View(result),即可查看列表中包含的内容。结果显示,其中包含了肿瘤表达数据TCGA_mat,细胞系表达数据CCLE_mat,以及两者分别的注释信息TCGA_ann和CCLE_ann。
三、计算基因表达平均值和方差
3.1 基因信息整理
common_genes<- intersect(colnames(result $TCGA_mat), colnames(result $CCLE_mat))
首先,使用intersect函数获取肿瘤表达数据TCGA_mat和细胞系表达数据CCLE_mat的共同表达基因。

hgnc.complete. set<- hgnc.complete. set%>% dplyr::select(Gene = symbol, Ensembl = ensembl_gene_id) %>% filter(Gene %in% common_genes) hgnc.complete. set<- hgnc.complete. set[!duplicated(hgnc.complete. set$Gene),] hgnc.complete. set<- hgnc.complete. set[common_genes,]
随后,根据得到的共同基因,对基因注释文件hgnc.complete.set进行相应的提取与整理。
3.2 计算基因表达平均值和方差
gene_stats < -data.frame( Tumor_SD= apply(result$TCGA_mat,2, sd, na.rm= T), CCLE_SD= apply(result$CCLE_mat,2, sd, na.rm= T), Tumor_mean= colMeans(result$TCGA_mat,na.rm= T), CCLE_mean= colMeans(result$CCLE_mat,na.rm= T), Gene= common_genes, stringsAsFactors= F)%> % dplyr::mutate(max_SD = pmax(Tumor_SD, CCLE_SD, na.rm=T)) gene_stats[1:3,]
使用apply函数,分别计算每个基因在

gene_stats <- left_join(hgnc.complete. set, gene_stats, by= "Gene") gene_stats[ 1: 3,]
使用left_join函数将计算得到的基因表达数据和前面整理好的基因注释文件进行合并。
结果显示:

write.table(gene_stats, file = "gene_expression_of_avg&SD.txt", sep = "\t",row.names = F,quote = F)
最后,将计算结果进行保存为txt文件,用于后续使用。
四、Figure 1A复现
接下来,开始正式的文章图表的复现过程。
4.1 表达数据和注释数据的准备
TCGA_mat<- result $TCGA_mat CCLE_mat <- result $CCLE_mat TCGA_ann <- result $TCGA_ann CCLE_ann <- result $CCLE_ann comb_ann <- rbind(TCGA_ann, CCLE_ann)
首先,分别提取表达数据和注释文件数据,并且使用rbind函数将肿瘤和细胞系的注释文件进行合并。
结果显示:

4.2 构建表达数据和注释信息的Seurat对象
original_combined_obj < -Seurat::CreateSeuratObject( t( rbind( TCGA_mat, CCLE_mat)), min.cells= 0,min.features= 0, meta.data= comb_ann%> % magrittr::set_rownames(comb_ann$sampleID))
使用Seurat包的CreateSeuratObject函数,其中min.cells参数表示至少在这么多数量的单元格中检测到特征;meta.data参数表示需要添加到
Seurat 对象的其他单元级注释数据,而且该注释信息应该是 data.frame形式,其中行是单元格名称,列是附加注释数据的内容。
结果显示:
original_combined_obj <- Seurat::ScaleData(original_combined_obj, features = rownames(Seurat::GetAssayData(original_combined_obj)), do.scale = F)
接着,将Seurat对象的表达值进行中心化处理。其中,这一过程的处理对后续PCA降维分析是十分关键的,它可以使数据具有统一的尺度(scale)。
结果显示:

4.3 PCA法降维
original_combined_obj %<>% Seurat::RunPCA(assay= 'RNA', features = rownames(Seurat::GetAssayData(original_combined_obj)), npcs = global$n_PC_dims, verbose = F)
使用RunPCA函数以对数据进行PCA 降维。
4.4 UMAP降维
original_combined_obj %<>% Seurat::RunUMAP(assay = 'RNA', dims = 1: global$n_PC_dims, reduction = 'pca', n.neighbors = global$umap_n_neighbors, min.dist = global$umap_min_dist, metric = global$distance_metric, verbose=F)
最后,使用非线性降维方法将这PCA分析得到的PC值降维到二维空间,其主要方法包括tSNE法或UMAP法。

4.5 结果可视化展示
uncorrected_alignment <- Seurat::Embeddings(original_combined_obj, reduction = 'umap') %>% as. data.frame %>% set_colnames(c( 'UMAP_1', 'UMAP_2')) %>% rownames_to_column( var= 'sampleID') %>% left_join(comb_ann, by= 'sampleID') colnames(uncorrected_alignment)
经过UMAP法降维分析后,最终得到UMAP_1和UMAP_2两个维度的数据。因此,将这两个维度的数据与注释信息根据样品名称sampleID进行匹配与合并,得到新的数据集,用于后续Figure 1A的输入数据。
结果显示:

uncorrected_combined_type_plot < -ggplot( uncorrected_alignment, aes( UMAP_1, UMAP_2)) + geom_point( data= filter(uncorrected_alignment,type== 'tumor'), alpha= 0.6,size= 0.5,pch= 21,color= 'white', aes( fill= type))+ geom_point( data= filter(uncorrected_alignment,type== 'CL'), alpha= 0.6,size= 0.6,pch= 3,aes( color= type),stroke= 0.5)+ scale_color_manual( values= c(CL= "#F8766D")) + scale_fill_manual( values= c(tumor= "#00BFC4")) + xlab(' UMAP1') + ggplot2::ylab(" UMAP2") + theme_classic+ theme( legend.position= 'bottom', text= ggplot2::element_text(size= 8), axis.text= ggplot2::element_text(size= 6), axis.title= ggplot2::element_text(size= 8), legend.margin= ggplot2::margin(0,0,0,0), legend.box.margin= ggplot2::margin(-10,-30,-10,-30), axis.line= ggplot2::element_line(size= .3))
以UMAP_1和UMAP_2两个维度的数据分别作为x轴和y轴进行绘制。结果显示,其中每个点代表一个组织样品或者一个细胞系,且红色代表细胞系,蓝色代表肿瘤组织。

这样,文章的Figure 1A就绘制完成了。
五、Supplementary Figure 1A复现
下面,我们来看下Supplementary Figure 1A的绘制过程。
5.1 Supplementary Figure 1A输入数据准备
library(sva)
考虑到Figure 1A中对表达数据的简单合并导致肿瘤组织和细胞系基本完全分离的情况。因此,我们需要对数据合并后进行去批次效应以对数据进行校正。在此,与GEO芯片数据合并过程一样,使用sva包来对数据的去批次效应。
dat <- rbind(TCGA_mat, CCLE_mat) cov_df <- comb_ann[match(comb_ann$sampleID, rownames(dat)), c( 'subtype', 'type', 'lineage', 'sampleID')] %>% dplyr::filter(!is.na(lineage), sampleID %in% rownames(dat)) dat <- dat[cov_df$sampleID, , drop = FALSE]
首先,根据注释分组信息将表达数据进行重新排序。
mod = model.matrix(~1, data=cov_df) combat_output < -sva::ComBat( t( dat), batch= cov_df$type,mod= mod) dim( combat_output)
根据两个数据集的相应信息,对参数batch和model进行设置,结合表达数据dat,将其导入到ComBat函数中,即可完成去批次操作。
结果显示:

5.2 Supplementary Figure 1A的绘制
combat_obj < -Seurat::CreateSeuratObject( combat_output, min.cells= 0, min.features= 0, meta.data= cov_df%> % magrittr::set_rownames(cov_df$sampleID)) combat_obj < -Seurat::ScaleData( combat_obj, features= rownames(Seurat::GetAssayData(combat_obj)), do.scale= F)
与前面的分析方法一样,通过构建新的Seurat对象,以及表达数据的中心化。
###PCA分析 combat_obj %<>% Seurat::RunPCA(assay= 'RNA', features = rownames(Seurat::GetAssayData(combat_obj)), npcs = global$n_PC_dims, verbose = F) ###UMAP分析 combat_obj %<>% Seurat::RunUMAP(assay = 'RNA', dims = 1: global$n_PC_dims, reduction = 'pca', n.neighbors = global$umap_n_neighbors, min.dist = global$umap_min_dist, metric = global$distance_metric, verbose=F)
接着,依次进行PCA分析和非线性UMAP降维分析。
###可视化展示 combat_res <- Seurat::Embeddings(combat_obj, reduction = global$reduction. use) %>% as.data.frame %>% magrittr::set_colnames(c( "UMAP_1", "UMAP_2")) %>% cbind.data.frame(cov_df[,c( "sampleID", "lineage", "subtype", "type")])
结果显示:
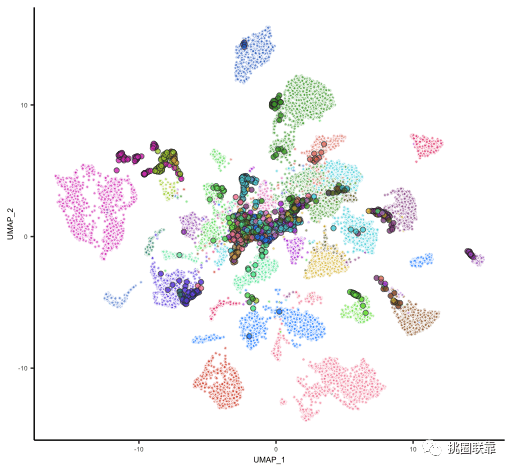
combat_plot < -ggplot( combat_res, aes( x= UMAP_1,y= UMAP_2,fill= lineage,size= type))+ geom_point( alpha= 0.6,pch= 21,color= "white") + geom_point( data= dplyr::filter(combat_res,type== "CL"), color= "#333333", pch= 21,alpha= 0.6)+ scale_size_manual( values= c(" CL" = 1.5," tumor" = 0.75))+ theme_classic+ theme( legend.position= "none", text= element_text(size= 6))+ ggplot2::scale_fill_manual( values= tissue_colors)
结果显示,其中小圆圈代表肿瘤组织,大圆圈代表细胞系,且不同的颜色代表了不同的组织类型来源。
到此,就已经完成了初步的数据整理,Seurat对象构建,以及结果的可视化展示。拿起你的数据,跟着一步步做下去,收获新的精美Figure。
转自:挑圈联靠
