承包你单细胞的所有美图_Scillus包
Hi,大家好,我是晨曦
今天这期推文来自一个小伙伴的留言
A同学:曦曦,除了ggplot2以外有没有更方便的方法可以更加丰富的展示我的scRNA-seq的结果,就是类似于scRNA-seq的ggpubr包
晨曦:有的,马上就来~
基于上面这个感兴趣的话题,我们就来看一下这个单细胞测序可视化的R包——Scillus包,可以承包你关于单细胞的所有美图哦~
那么我们开始吧
首先我们来给Scillus包一个精炼的定义:Seurat wrapper for enhanced processing and visualization of scRNA-seq data
其含义就是针对Seurat对象的可视化增强器(装备感满满~,感觉好像可以一刀9999)
当然了,前提是我们已经掌握了使用Seurat进行单细胞分析的标准流程,然后我们就可以使用Scillus包对我们的结果进行更为丰富的可视化展示
那么我们接下来就开始进行代码实操
#安装R包 if (!require(devtools)) { install.packages("devtools") } devtools::install_github("xmc811/Scillus", ref = "development")
晨曦解读
作者把单细胞处理和可视化都进行了封装,通过运行确实是一个对于自己单细胞分析过程的复习,而且数据集比较少, 推荐大家也跑跑看,但是如果你已经有Seurat对象了,这里就可以直接进行后续的可视化
#加载R包 library(Scillus) library(tidyverse) library(Seurat) library(magrittr) #导入数据 a <- list.files("可视化/GSE128531_RAW", full.names = TRUE)#获取数据集列表 #六个样本的单细胞数据(每一个样本都是标准的三个文件) m <- tibble(file = a, sample = stringr::str_remove(basename(a), ".csv.gz"), group = rep(c("CTCL", "Normal"), each = 3))#获取数据集列表的表格形式并且提供分组信息 pal <- tibble(var = c("sample", "group","seurat_clusters"), pal = c("Set2","Set1","Paired"))#可视化调色盘 #读取表达数据并自动创建Seurat对象 scRNA <- load_scfile(m) length(scRNA) #[1] 6
Scillus 将为每个样本创建 Seurat 对象,并自动调用 PercentageFeatureSet函数来计算线粒体基因百分比。由此产生多个Seurat对象的列表,它的长度等于元数据 m 的行数
plot_qc(scRNA, metrics = "percent.mt")#展示每个样本的线粒体基因比例
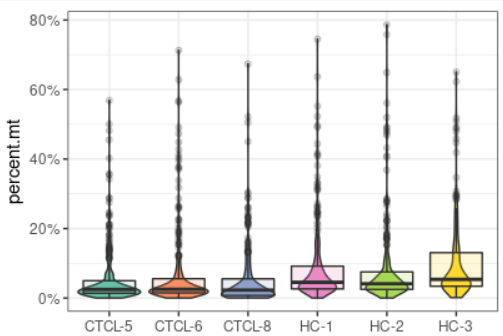
plot_qc(scRNA, metrics = "nFeature_RNA")#展示每个样本基因的数量
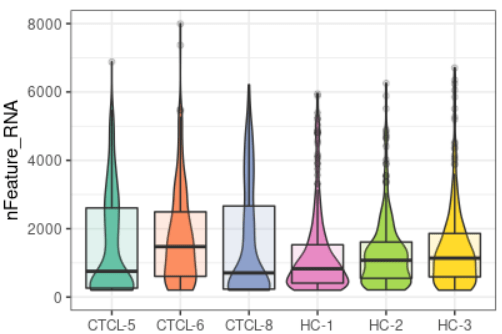
plot_qc(scRNA, metrics = "nCount_RNA")#展示每个样本mRNA的数量
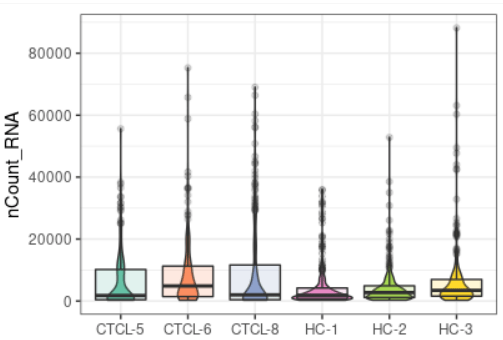
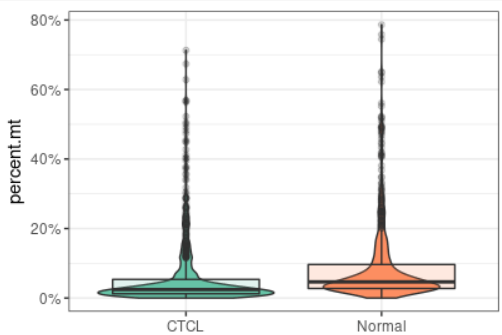
plot_qc(scRNA, metrics = "percent.mt", plot_type = "box")#展示箱线图的线粒体基因比例
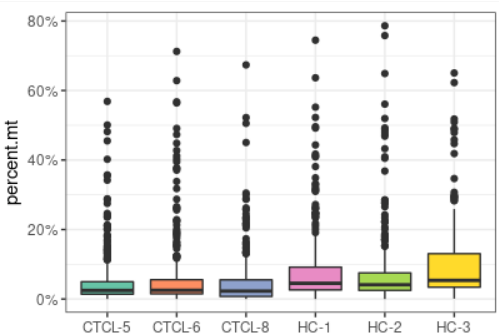
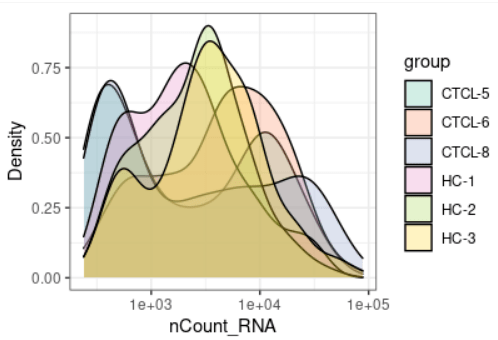
plot_qc(scRNA, metrics = "nCount_RNA", plot_type = "density") + scale_x_log10 #展示mRNA的密度图
plot_qc(scRNA, metrics = "percent.mt", group_by = "group")#展示不同分组下线粒体基因的比例 #这块的思路晨曦觉得作者做的真的很巧妙 #我们前面是通过m变量数据批量构建的Seurat对象,m变量如下 # A tibble: 6 × 3 # file sample group # <chr> <chr> <chr> #1 可视化/GSE128531_RAW/CTCL-5.csv.gz CTCL-5 CTCL #2 可视化/GSE128531_RAW/CTCL-6.csv.gz CTCL-6 CTCL #3 可视化/GSE128531_RAW/CTCL-8.csv.gz CTCL-8 CTCL #4 可视化/GSE128531_RAW/HC-1.csv.gz HC-1 Normal #5 可视化/GSE128531_RAW/HC-2.csv.gz HC-2 Normal #6 可视化/GSE128531_RAW/HC-3.csv.gz HC-3 Normal #通过m变量这样导入生成Seurat对象天生就附带了元数据的特性也就是大分组信息,保留在seurat对象中的是是样本信息scRNA[[1]]@project.name
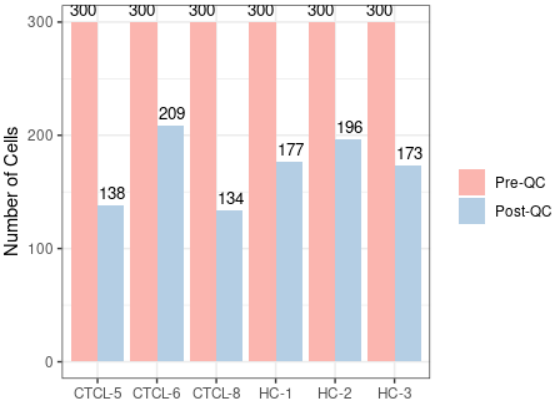
#当然除了上面还有一些更多展示QC方式的可视化,代码如下,各位小伙伴可以私下运行学习 plot_qc(scRNA, metrics = "percent.mt", group_by = "group", pal_setup = "Accent") plot_qc(scRNA, metrics = "percent.mt", group_by = "group", pal_setup = pal) plot_qc(scRNA, metrics = "percent.mt", group_by = "group", pal_setup = c("purple","yellow")) #基于Seurat的标准piplines scRNA_f <- filter_scdata(scRNA, subset = nFeature_RNA > 500 & percent.mt < 10) #并且自动显示质控结果(这个功能感觉超棒)
scRNA_f %<>% purrr::map(.f = NormalizeData) %>% purrr::map(.f = FindVariableFeatures) %>% purrr::map(.f = CellCycleScoring, s.features = cc.genes$s.genes, g2m.features = cc.genes$g2m.genes) scRNA_int <- IntegrateData(anchorset =
FindIntegrationAnchors(object.list = scRNA_f, dims = 1:30, k.filter =
50), dims = 1:30) scRNA_int %<>% ScaleData(vars.to.regress = c("nCount_RNA", "percent.mt", "S.Score", "G2M.Score")) scRNA_int %<>% RunPCA(npcs = 50, verbose = TRUE) #%>% 管道符号不赋值就只是给你看看 #%<>% 结果自动进行赋值 scRNA_int %<>% RunUMAP(reduction = "pca", dims = 1:20, n.neighbors = 30) %>% FindNeighbors(reduction = "pca", dims = 1:20) %>% FindClusters(resolution = 0.3)
简单来说,上面这个piplines就可以帮助我们完成scRNA-seq多数据集联合,而且重点是这个piplines是可以复制的
#我们把元数据的分组信息提取出来,方便我们后续作图 m %<>% mutate(group = factor(group, levels = c("CTCL","Normal"))) scRNA_int %<>% refactor_seurat(metadata = m)
至此,我们的数据处理过程就结束了,这个流程可以复制到我们scRNA-seq数据多数据集联合上,当然我们标准的Seurat对象标准流程也是可以使用后续的可视化方式
注意:如果不是基于这个R包获得的对象,尽管对象本质都是一样,但是有些信息是缺少的,可能只能可视化部分图表,需要额外添加信息
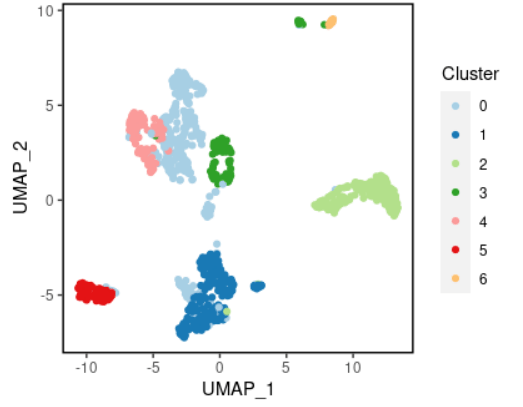
#可视化案例1 plot_scdata(scRNA_int, pal_setup = pal)
#可视化案例2 plot_stat(scRNA_int, plot_type = "group_count")
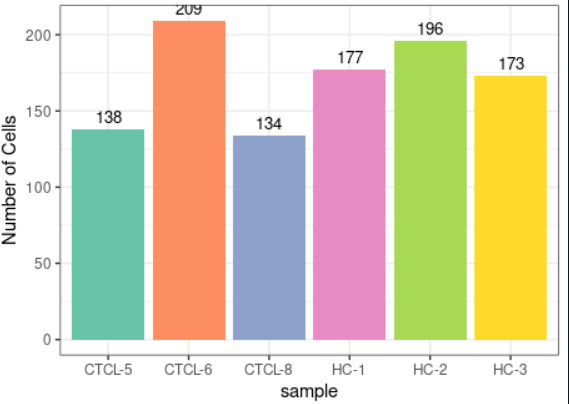
#可视化案例3 markers <- FindAllMarkers(scRNA_int, logfc.threshold = 0.1, min.pct = 0, only.pos = T) plot_heatmap(dataset = scRNA_int, markers = markers, sort_var = c("seurat_clusters","sample"), anno_var = c("seurat_clusters","sample","percent.mt","S.Score","G2M.Score"), anno_colors = list("Set2", # RColorBrewer palette c("red","orange","yellow","purple","blue","green"), # color vector "Reds", c("blue","white","red"), # Three-color gradient "Greens"))
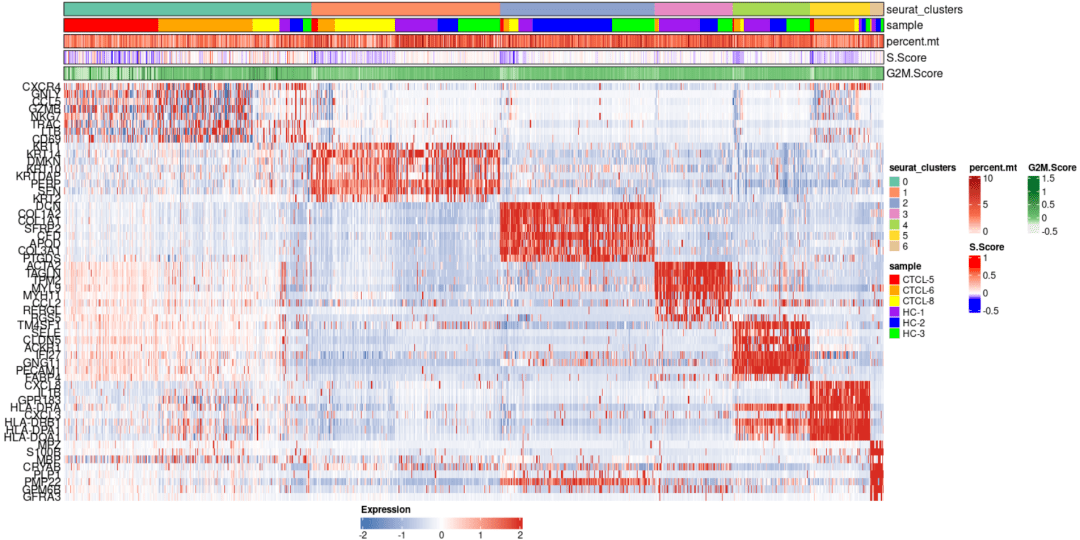
#可视化案例4 plot_cluster_go(markers, cluster_name = "1", org = "human", ont = "CC")
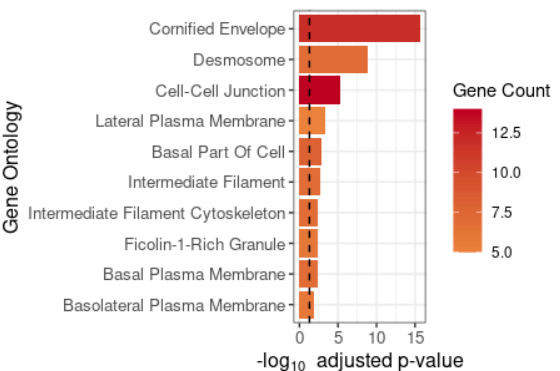
#可视化案例5 de <- find_diff_genes(dataset = scRNA_int, clusters = as.character(0:6), comparison = c("group", "CTCL", "Normal"), logfc.threshold = 0, # threshold of 0 is used for GSEA min.cells.group = 1) # To include clusters with only 1 cell gsea_res <- test_GSEA(de, pathway = pathways.hallmark) plot_GSEA(gsea_res, p_cutoff = 0.1, colors = c("#0570b0", "grey", "#d7301f"))
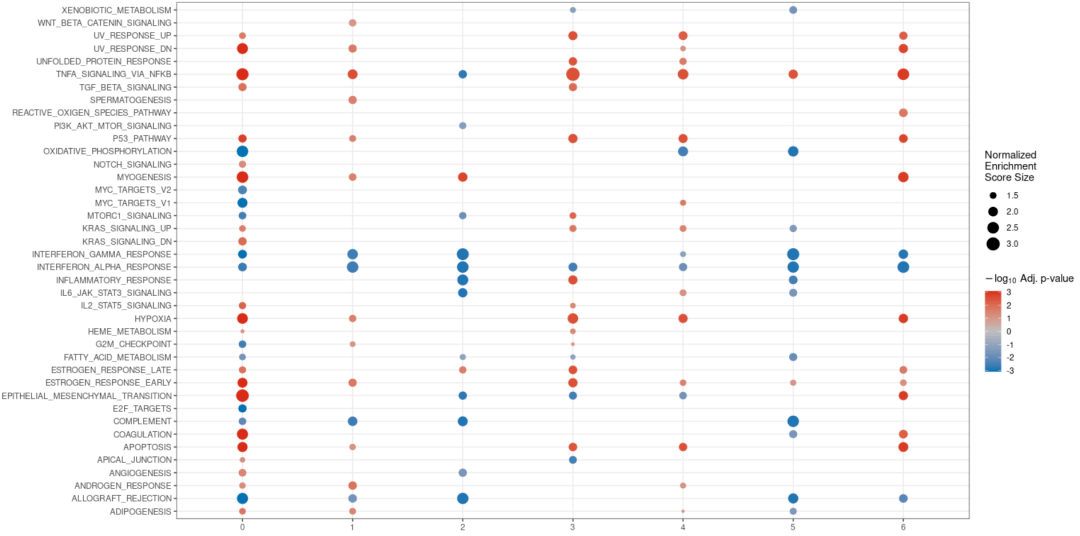
当然还有其它可视化的结果,这里晨曦就不一一展示了,各位小伙伴可以查看下面的R包地址进行进一步的学习:Home | Scillus
这个R包晨曦使用最大的亮点我觉得有三点:
1. 提供了一个多数据集联合的piplines,可复制性极强
2. 优化了Seurat的可视化展示(其中的复杂热图很好看~)
可视化的展示每一种都可以展示亚群、分组以及基因和样本这些基本的Seurat可以可视化的信息
3. 提供了GO和GSEA的一键式分析
那么,本期推文到这里就结束啦~
我是晨曦,我们下期再见
