这一节里,我们来看一下独立资源的几个例子。
一个很重要的资源是蛋白三维结构数据库 Protein DataBank (PDB)。我们这门课里对于蛋白三维结构的计算和预测讲的很少,但这其实也是非常重要的资源和研究方向。Protein DataBank现在主要是由Rutgers和UC Sandiego共同来管理。
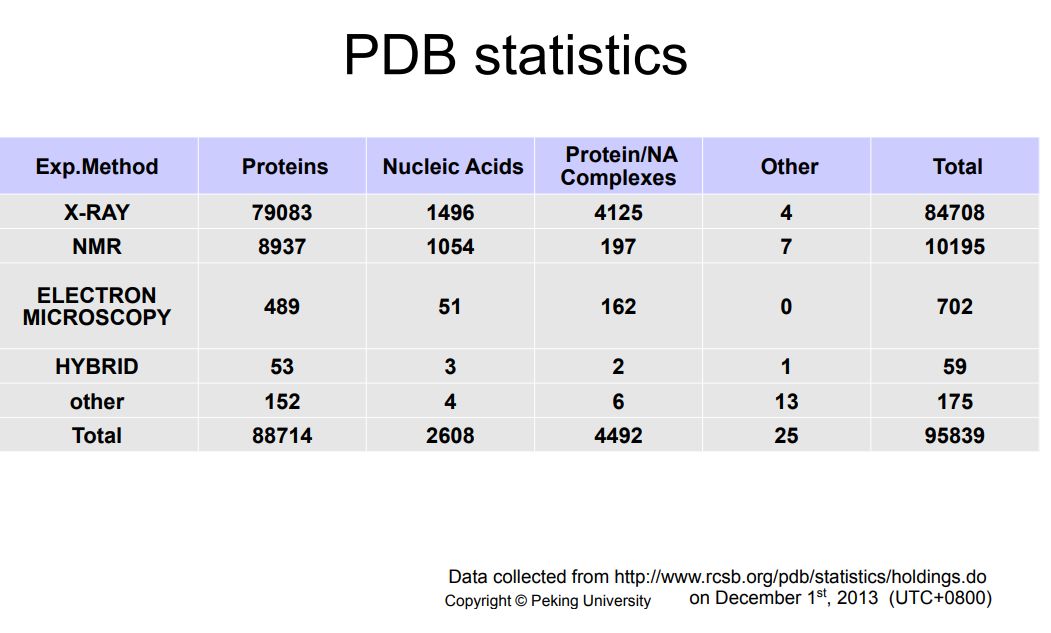
它目前共有9万多,将近10万的三维结构,包括8万多的X-ray晶体结构,1万多的核磁结构,还有其它的像Electron Microscopy等等的一些结构。
这些结构中最多的是蛋白的结构,另外也有核酸结构,和蛋白和核酸的复合体的结构。
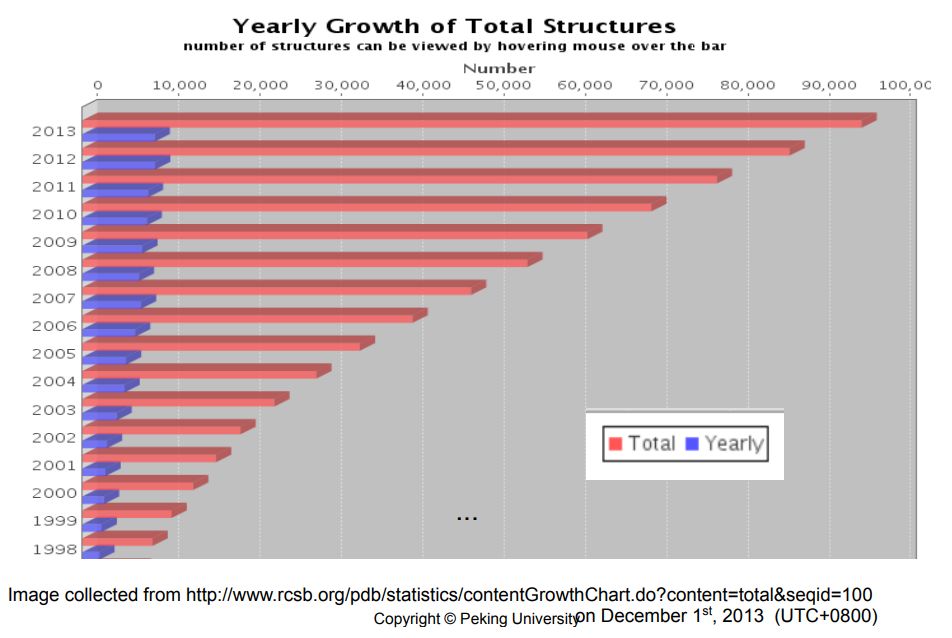
解蛋白结构不像测序这样能够有这么高的通量。这个图显示的是PDB这么些年来增长的一个趋势。 蓝的显示的是每一年新增加的蛋白三维结构的数量,每一年都有增加,但增加的不是那么多。但是要是看总数的话,还是有一个比较大的增加。这里90%是蛋白的结构,10%是核酸和复合体的结构。
Stanford有一个比较有用的资源叫GERP,它主要是来计算保守性。你如果给它一组蛋白序列,它就可以找到这一组序列里最保守的区间是什么。
再举另外的一个例子。这是Yale的Gerstein Lab做的CNVnator。它主要是通过新一代测序技术来鉴定拷贝数变异。 类似的还有像BICseq等等的几个相关的这种软件。

Sanger Institute做了一个Rfam这样的一个数据库及相关的工具,把已知的很多非编码RNA——非编码RNA它自己也有很多的家族——的家族做了比对和相应的motif[分析]。 然后如果你有一个新的非编码RNA测量出来,你想知道它有什么潜在的功能,你就可以用这个Rfam来做一个预测。

最后给大家简单介绍一下还有一类很重要的资源,就是我在第一节里也简单提到过的这些小的程序包,包括最主要的就是Bioconductor,BioPerl,和BioPython。
Bioconductor里有很多小的R包。一般来说,在你动手写一个R程序之前,你要先到Bioconductor去看一下有没有别人写了。 别人写的程序你也不能直接拿过来不动脑子地用,你也要看它的程序,你要做测试,确认它没有问题。
不要到时候被别人一个错误的程序把你给害了。这个资源是非常非常的有用的,而且[它是]随着时间的推移会越来越重要的一个资源。
类似的一个资源是BioPerl,[它]有很多Perl的程序包。另外它可以装到Linux、Windows、Macintosh等等很多不同的平台。它也有很多的帮助文档,比如说How do I learn Perl?
BioPython就是类似的。R语言、Perl、Python也是生物信息学用的最多的三种编程语言。 如果是计算量很大的一些程序,我们都还是要用C和C++来写。但是其它的很多程序,尤其是[把]你已知的几个程序包裹一下的话,用的最多的就是Perl和Python。而涉及到统计分析,用的很多的就是R。

所以像我前面提到的,生物信息学发展的很快,有很多没有解决的问题,[是]一个非常年轻很有活力的一个领域。 两年之后我们再上这门课,可能就会再讲新的内容,[其中就]可能有在坐的某一个同学开发的一个工具。另外,即使你自己不开发,[也]一定要去用。这是生命科学必然的一个趋势。一定是很重要的一部分。

[这里要]提醒大家,像Spider-Man电影里[那样说的],不知道大家有没有记得,Spider-Man电影里第一集有很著名的一句话,叫”With great power comes great responsibility.” 我觉得[这句话无论]对于Spider-Man这种power还是对于一个实验技术都是适用的。尤其像生物信息学的方法,它有一个特点,就是不管你输入什么数据,你一click、run,它都会给你个结果。
如果你对这个方法的底层的假设、局限性、准确性不了解的话,你很有可能就做出一个很错误的推断;如果你对它很了解的话,这真的就是一个很有用的power。
一分钟抓住一个知识点
(CNV)Copy-number variation
拷贝数变异(CNV)是一种基因组片段重复的现象,基因组中重复的数量因个体而异。拷贝数变化是一种结构变化:具体来说,它是一种影响大量碱基对的复制或删除事件。然而,请注意,尽管现代基因组学研究主要集中在人类基因组上,复制数的变化也发生在其他多种生物中,包括大肠杆菌和酿酒酵母。
最近的研究表明,大约三分之二的人类基因组由重复的序列组成,4.8-9.5%的人类基因组可分为拷贝数变异。在哺乳动物中,拷贝数的变化在种群和疾病表型的必要变化中起着重要的作用。
拷贝数变化一般可分为两大类:短重复和长重复。然而,这两个群体之间没有明确的界限,分类取决于感兴趣的位点的性质。短重复主要包括双核苷酸重复(两个重复的核苷酸,如A-C-A-C-A-C…)和三核苷酸重复。长重复包括整个基因的重复。这种基于重复大小的分类是最明显的分类类型,因为大小是检查最有可能产生重复的机制类型的一个重要因素,因此这些重复对表型的可能影响。
——维基百科

转自华康基因
- 本文固定链接: https://maimengkong.com/zu/1393.html
- 转载请注明: : 萌小白 2023年3月4日 于 卖萌控的博客 发表
- 百度已收录
