微生物组测序 (主要指扩增子测序、全长扩增子测序与宏基因组测序)可提供细菌构成、基因丰度和功能性信息,可以解决“who is there”(那儿有谁)和“what are they doing”(在干嘛)的问题。而代谢组学是研究生物体中代谢产物变化的科学,可以解决“what have really happened”(究竟发生了什么)的问题。生物科学研究过程复杂,单独和片面的单一组学无法解释清楚生物学问题,多组学就显得尤为重要。近年来,随着微生物组学研究的不断发展和持续火热,越来越多的研究者开始将微生物组学和代谢组学联合起来,从物种、基因以及代谢产物等水平共同解释科学问题,更好地理解疾病病变过程及机体内物质的代谢途径。还有助于发现疾病的生物标记以更进一步应用于临床辅助诊断。代谢和微生物的联合分析可以有多种方法,今天主要介绍相关性分析和协惯量分析。
文章《Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention》中揭示了中国肥胖人群的肠道菌群组成,并指出了肥胖及减肥干预后肠道菌群和血清代谢物改变。其中MLG(metagenomic linkage groups,宏基因组连锁群)和代谢物联合分析就运用了相关性分析和协惯量分析(如下图)。

这里使用R基于示例数据绘制类似的图,需要提前安装psych、ade4、pheatmap等R包。另外使用TakeColor软件可直接获取文章图中的配色。使用的示例数据为6个样本的数据,对照处理各3个样本。
#代谢组数据,行为代谢物共20个,列为样本名
> dim(metabdata)
[1] 20 6
# 微生物数据,行为微生物共47个,列为样本名
> dim(microdata)
[1] 47 6
# 加载相关性计算的包
> library(psych)
# 使用spearman计算相关性和p值
> cor_res<-corr.test(cbind(t(metabdata),t(microdata)),method= 'spearman',adjust= 'none',ci=F)
# 显示数据
> dim(cor_res $p)
[1] 67 67
> tail(cor_res $r[,1:3])
Cyclic AMP Tricin Genistein
Pseudomonas 0.7714286 0.9428571 -0.7714286
Ralstonia 0.6667367 0.8406680 -0.8986451
Rhizobium 0.8285714 0.6571429 -0.7142857
Saccharopolyspora 0.8406680 0.6667367 -0.7247138
Terriglobus 0.8986451 0.7247138 -0.6667367
Vicinamibacter 0.7142857 0.8857143 -0.8285714
# 提取相关性和p值
> corCmat<-cor_res $r[(nrow(metabdata)+1):nrow(cor_res $r),1:nrow(metabdata)]
> corPmat<-cor_res $p[(nrow(metabdata)+1):nrow(cor_res $p),1:nrow(metabdata)]
# 绘制热图
> library(pheatmap)
> library(RColorBrewer)
# 标记显著当p<0.01 显示2个星号‘**’,0.01<p<0.05 时显示一个星号‘*’
> annolabel<-matrix( '',nrow(corPmat),ncol(corPmat))
> annolabel[corPmat<0.01]<- '**'
> annolabel[corPmat<0.05 & corPmat>=0.01]<- '*'
> colnames(annolabel)<-colnames(corPmat)
> rownames(annolabel)<-rownames(corPmat)
pheatmap(mat=corCmat,
display_numbers=annolabel,
number_color='black',
cluster_cols=T,
cluster_rows=T,
color=colorRampPalette(c('#7AA1D3','#D2609E'))(100),
border_color=F,
show_rownames=T,
show_colnames=T,
fontsize_number=8,
fontsize_row=6,
fontsize_col=6,
scale='none')
(向左查看更多内容)
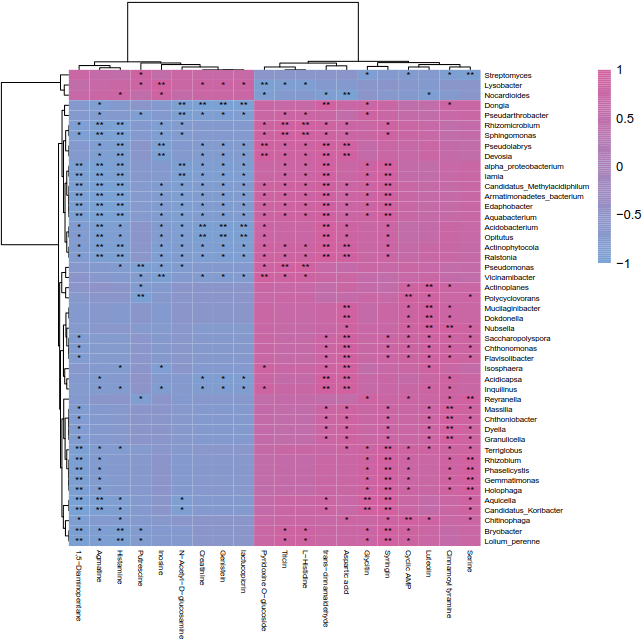
协惯量分析:
#加载包
> library(ade4)
#进行协惯量分析
> microdata_dudi <- dudi.pca(t(microdata), scale = TRUE, scan = FALSE, nf = 2)
> metabdata_dudi <- dudi.pca(t(metabdata), scale = TRUE, scan = FALSE, nf = 2)
> coin<- coinertia(metabdata_dudi,microdata_dudi, scan = FALSE, nf = 2)
#基于特征值计算贡献
>Axis1<-coin$eig[ 1]/sum(coin$eig)* 100
>Axis2<-coin$eig[ 2]/sum(coin$eig)* 100
#绘制样本空间图,展示样本之间的差异
>metabS<-coin$mX
>microS<-coin$mY
>plot(microS[, 1],microS[, 2], xlim = c( -1.5, 1.5), ylim = c ( -2, 2),
pch= 20,col=c(rep( '#4890CD', 3),rep( '#EC2B91', 3)),
xlab=sprintf( 'Axis1(%.2f%%)',Axis1),ylab=sprintf( 'Axis2(%.2f%%)',Axis2))
>arrows(x0=microS[, 1],y0=microS[, 2],
x1=metabS[, 1],y1=metabS[, 2],
col=c(rep( '#4890CD', 3),rep( '#EC2B91', 3)),length= 0.1)
(向左查看更多内容)
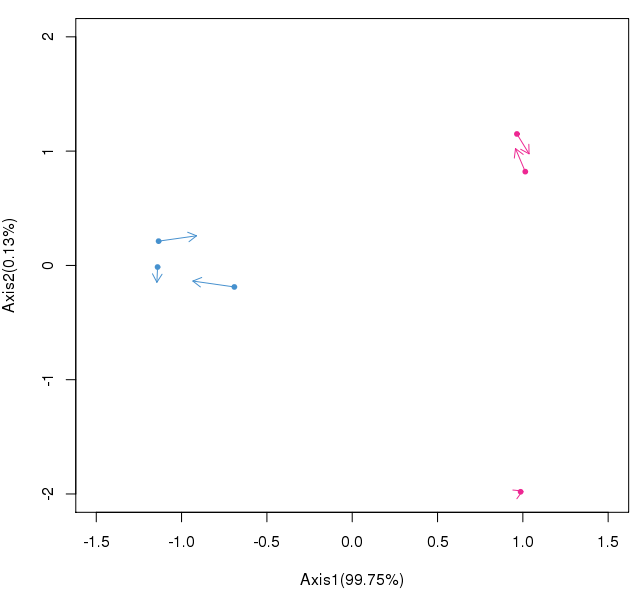
图中的蓝色和红色分别代表不同的样本分组即对照和处理,基于此分析可直观展示出样本间的差异。
对于没有R基础的同学可能会觉得困难,但是没关系,目前百迈客云已有代谢组与微生物联合分析以及个性化相关的绘图,只需要点点鼠标便可得到结果。
文: QG
排版:市场部
参考文献:
Liu R, Hong J, Xu X, et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention[J]. Nature medicine, 2017, 23(7): 859.
转自:百迈客生物- 本文固定链接: https://maimengkong.com/kyjc/1049.html
- 转载请注明: : 萌小白 2022年6月27日 于 卖萌控的博客 发表
- 百度已收录
