为什么pathway富集分析结果没有我感兴趣的通路?
GO和KEGG富集分析使用差异基因(上调基因,下调基因,或者上下调合起来的基因)作为输入,使用超几何分布等算法计算显著富集的GO term或者通路,然而,在实际数据处理中,这种使用p值和fold change进行一刀切获得差异基因,然后进行富集分析的分析方法,往往富集不到我们感兴趣的结果。这时,可以试试基因集富集分析(Gene set enrichment analysis,GSEA),它使用全部基因作为输入,找出具有协同差异 (concordant differences)的基因集,兼顾了差异较小的基因(因为在某些条件下,1.5倍的差异可能就算很大的了)。因此,Broad institute出品的GSEA在论文中应用广泛。
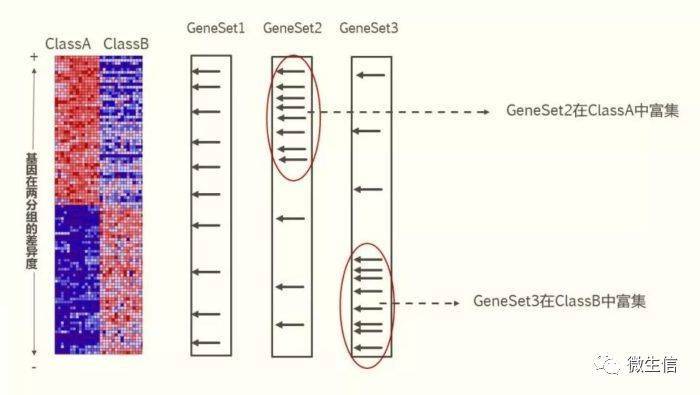
图1 GSEA原理(图片来自plob.org)
GSEA分析原理
1,基因排序:
利用所有基因的表达值,计算每个基因在两个表型(ClassA和ClassB)间的差异程度(GSEA提供了6种算法,默认是signal2ratio),然后按照差异程度将基因从大到小排序。这里差异是有正有负的,正值越大表示在ClassA (vs ClassB)中表达越高,越负表示在ClassA中表达越低。
2,分析基因集是否富集: 基因集(gene set )是一类具有相关功能(例如免疫相关)或者符合某一标准(例如某个miRNA的2 00 个靶基因)的基因构成的一组基因。 图中GeneSet1(一个箭头代表一个基因)里的基因在排序的基因列表里均匀分布(表明这个基因集不在这两个表型中富集),GeneSet 2 里的基因主要分布在基因列表的顶部(表明在 C lassA中富集),GeneSet3里面的基因主要分布在基因列表的底部(表明在Class B 中富集)。 3,计算富集分数: 计算每个基因集的富集分数(enrichment score,ES),然后对ES分数进行显著性检验及多重假设检验,从而计算出显著富集的基因集。常见的GSEA分析软件及评测:
目前常见的gsea分析软件包括:
官方Broad的GSEA;
R版的fgsea,clusterprofiler;
Python版的GSEApy等
Broad GSEA软件分析的两种模式 1,常规模式: 输入表达矩阵,软件自动计算foldchange,由于要进行显著性检验,因此至少3vs3。 2,Prerank模式: 输入排序后的基因列表,针对那些例如1vs1这种不能用常规模式计算的数据。我们使用同一套数据集测试了Broad GSEA prerank模式,ClusterProfiler,GSEApy发现:
1,Broad GSEA最慢,ClusterProfiler最快,算法不太一样
2, Broad GSEA结果与GSEApy的结果重叠度最高,而与clusterProfiler结果重叠度最低。当然三个结果总体上还是趋于一致的。
3, GSEApy占内存最大,可以多线程
4, ClusterProfiler可以绘制多个富集结果,Broad GSEA和GSEApy不可以
5, GSEApy可以添加相关的NES,Pvalue值等,Broad GSEA和clusterProfiler不可以 6, 出图美观度:个人感觉GSEApy稍微好看些
基于以上测试结果,经过权衡,我们上线了基于ClusterProfiler的基因集富集分析页面。

图2. GSEA输出示例
1 , 1,打开GSEA分析和绘图页面
首先,使用浏览器(推荐chrome或者edge)打开GSEA分析和绘图页面。左侧为常见作图导航,中间为数据输入框和可选参数,右侧为描述和结果示例。也可以在主页搜索框中搜索gsea,找到gsea分析和绘图页面。
https://www.bioinformatics.com.cn/plot_basic_gene_set_enrichment_analysis_gsea_analysis_193
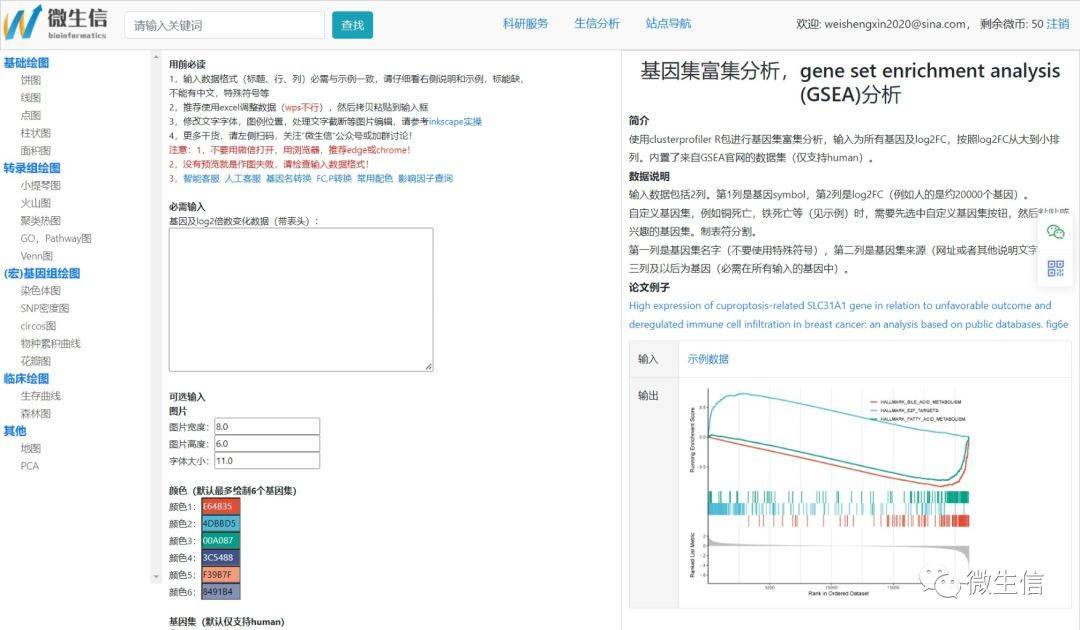
图3.GSEA分析页面
2,示例数据
点击右侧“示例数据”链接下载excel格式的示例数据。
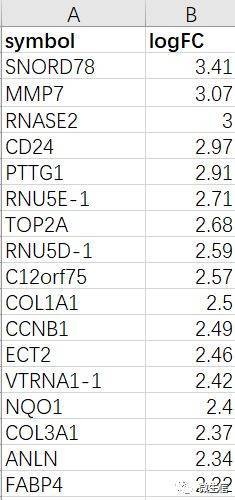
图4. 输入数据示例
示例数据(仅供参考)包括2列:
第1列是基因名(symbol)
第2列是倍数变化(从大到小排列,正的在顶部,负的在底部)
注意:这里是全部的基因(例如人的约2w个基因)
3,粘贴示例数据
直接复制示例数据中的AB两列数据,然后粘贴到输入框。
注意:不是拷贝excel文件,是拷贝excel文件里边的数据。另外粘贴到输入框后,格式乱了没关系,只要在excel中是整齐的就行。并且数据矩阵中不能有空的单元格,中文字符等。
图5. 必需输入
4,修改参数,并提交
我们设置了图片尺寸,文字大小,颜色等参数,并内置了GSEA官网(http://www.gsea-msigdb.org/gsea/msigdb/index.jsp)的多个基因集,包括最常用的:hallmark基因集,kegg基因集等(这些基因集仅支持human)

图6.可调参数
5,提交分析
粘贴好输入数据,调整好参数(或者全部默认)后,点击提交按钮,约15秒后,会在页面右侧出现富集结果预览图和分析结果。我们提供了4种图片格式供下载使用,两种矢量图(pdf,svg)和两种标量图(600 dpi tiff和300 dpi png)。
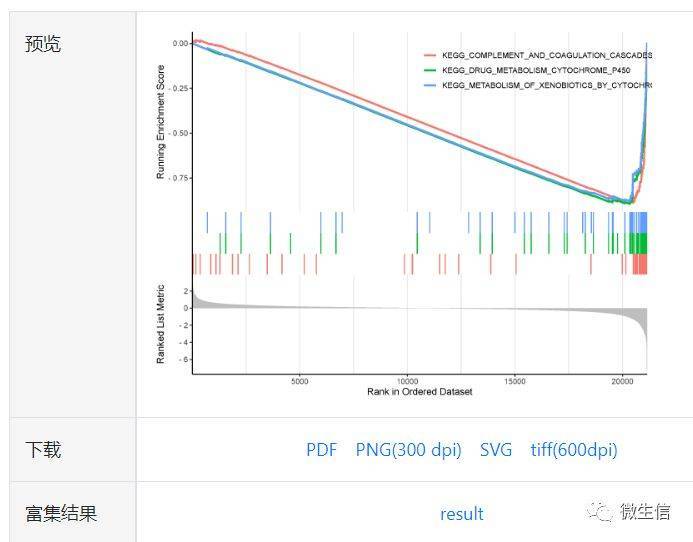
图7.预览与下载
结果解析
该图分为3块:
最上方:Enrichment Score折线图。横坐标是排序后的基因,纵坐标是对应的Running ES, 折线的峰值是这个基因集的富集分数(Enrichment Score,ES)。正值说明在ClassA中富集,峰值左边的基因为核心基因,负值相反(见原理)
中间:基因集中基因在基因排序列表中所处的位置,也就是将图1中的三个垂直数据集转动了90度摆放。如果所研究的基因集中的基因显著聚集在左侧,则说明该基因集与ClassA相关,显著富集在右侧,说明与ClassB相关。
下方:每个基因对应的ranked list metric,以灰色面积图展示。
结果文件如下:
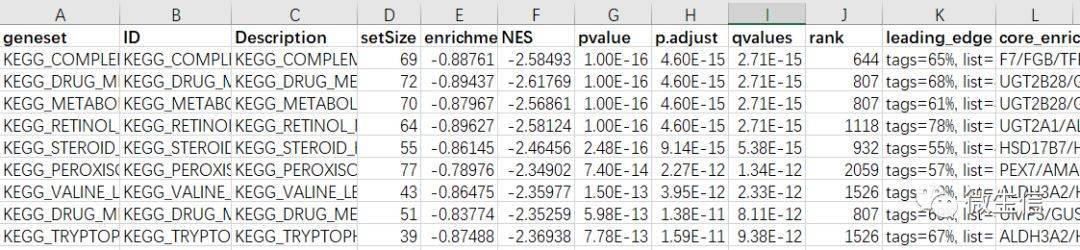
图8. Gsea分析结果
各列说明:
Geneset, ID,Description:基因集名字及描述
SetSize:富集到该基因集的基因个数
Enrichment score:富集分数ES
NES:标准化的ES,normalized enrichment score
Pvalue:富集的p值
p.adjust:校正p值
qvalues:qvalue
rank:排名
core_enrichment:富集到该通路的核心基因列表
一般来说:NES绝对值越大,FDR值越小,说明富集程度越高,结果越可靠。
重点来了,自定义数据集
常规的GSEA仅支持human物种,因此在对非human物种进行GSEA分析时,我们首先需要定义一个基因集,这个基因集可以来自文献,数据库等。以细胞焦亡、铜死亡、铁死亡等基因集为例,首先选择自定义基因集按钮,然后将相关基因按照自定义基因集示例格式贴到自定义输入框:
一行一个基因集。第一列是名字,第二列是来源,后续列为该基因集里边的基因,尽量避免使用特殊符号,并且这些基因名必需在你输入的全部基因里。
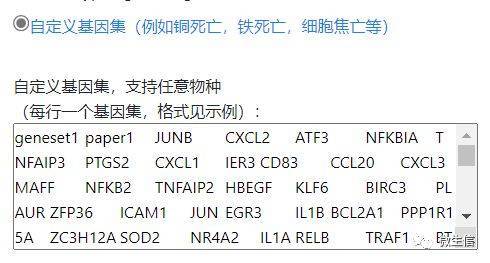
图9. 自定义基因集
点击提交按钮,约15s后,会在右侧出现自定义基因集的富集结果。
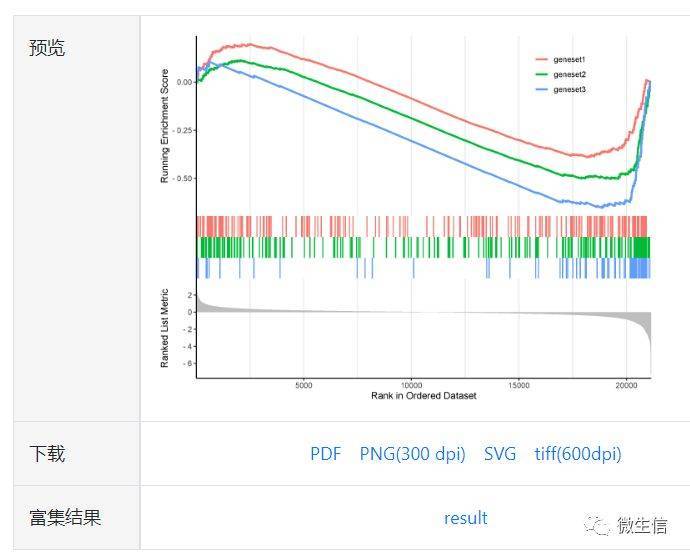
图10. 自定义基因集输出结果
想看你的数据是否跟最新的铜死亡,铁死亡,锌死亡,细胞焦亡等热点相关,可以先拿这些基因集跑个GSEA试试看,这就是自定义基因集的强大之处。
没有预览就是没有出图,这时请参考示例数据,检查自己输入数据的格式。
遇到文字截断,需要修改字体、调整字体大小等,使用scape软件。
- 本文固定链接: https://maimengkong.com/image/1248.html
- 转载请注明: : 萌小白 2022年10月6日 于 卖萌控的博客 发表
- 百度已收录
