

2024
02-06
02-06
经验分享:非编码RNA(non-coding RNA, ncRNA)简介
 非编码RNA(non-coding RNA, ncRNA)是一种独特的RNA转录本,占人类基因组RNA的90%以上,除少数具有开放阅读框,因而具有编码潜能以外,通常不编码蛋白质,而是作为发育、增殖、转录、转录后修饰、凋亡、细胞代谢等多种生物过程的重要调控因子。广义上讲,转运 RNA ,核糖体 RNA ,核内小分子 RNA 及核酶等都属于非编码 RNA, 对于这些非编码 RNA 的结构和功...阅读... 阅 读 全 部 >
非编码RNA(non-coding RNA, ncRNA)是一种独特的RNA转录本,占人类基因组RNA的90%以上,除少数具有开放阅读框,因而具有编码潜能以外,通常不编码蛋白质,而是作为发育、增殖、转录、转录后修饰、凋亡、细胞代谢等多种生物过程的重要调控因子。广义上讲,转运 RNA ,核糖体 RNA ,核内小分子 RNA 及核酶等都属于非编码 RNA, 对于这些非编码 RNA 的结构和功...阅读... 阅 读 全 部 >
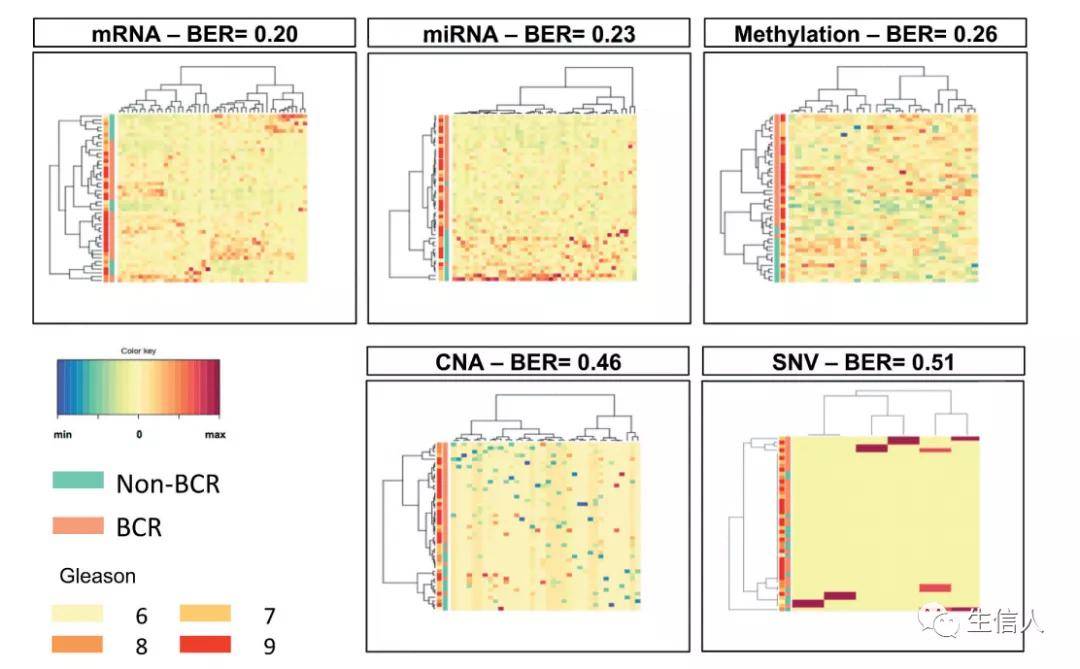 今天小编要和大家分享的是去年12月发表在OncoImmunology(IF:5.869)杂志上关于免疫聚焦多组学数据分析前列腺癌的文章,更多生信人原创文章见:生信人。https://biosxr.gaptools.cn/#/Immune-focused multi-omics analysis of prostate cancer: leukocyte Ig-Like receptors...阅读...
今天小编要和大家分享的是去年12月发表在OncoImmunology(IF:5.869)杂志上关于免疫聚焦多组学数据分析前列腺癌的文章,更多生信人原创文章见:生信人。https://biosxr.gaptools.cn/#/Immune-focused multi-omics analysis of prostate cancer: leukocyte Ig-Like receptors...阅读...  人类基因组计划自启动以来,目前已经出了很多版本的人类基因组,例如hg18, hg19, GRCh37, GRCh38。为了将不同版本的基因组位置信息一一对应,UCSC 推出了一个基于基因组序列的转换工具LiftOver。它能够快速准确的将不同版本的基因组坐标进行相互转换,已被广泛应用于各种基因组研究之中。UCSC分别提供了网页版和Linux命令行两个版本的LiftOver工具。网...阅读全文&g...
人类基因组计划自启动以来,目前已经出了很多版本的人类基因组,例如hg18, hg19, GRCh37, GRCh38。为了将不同版本的基因组位置信息一一对应,UCSC 推出了一个基于基因组序列的转换工具LiftOver。它能够快速准确的将不同版本的基因组坐标进行相互转换,已被广泛应用于各种基因组研究之中。UCSC分别提供了网页版和Linux命令行两个版本的LiftOver工具。网...阅读全文&g... 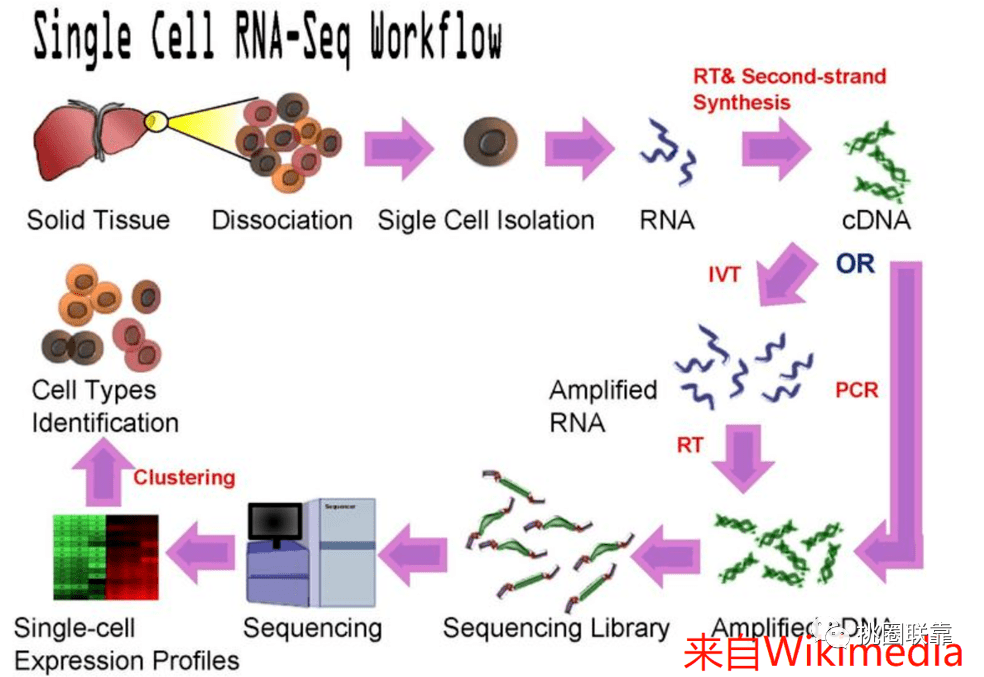 高质量scRNA-seq数据检索/注释工具:Cell BLAST 嗨,小伙伴们大家好!哈哈~新的一年祝大家牛年大吉,多发paper,多拿基金,工作科研两开花,家庭事业双福气!为配合风哥单细胞测序分析R语言全代码系列,知你所求懂你所需,本着饭喂到嘴边的少妇原则,弘毅给大家带来相关的无代码数据库介绍。这周就从Cell BLAST开始,一款高质量scRNA-seq数据检索/注释工具,一起来康康吧~! ...
高质量scRNA-seq数据检索/注释工具:Cell BLAST 嗨,小伙伴们大家好!哈哈~新的一年祝大家牛年大吉,多发paper,多拿基金,工作科研两开花,家庭事业双福气!为配合风哥单细胞测序分析R语言全代码系列,知你所求懂你所需,本着饭喂到嘴边的少妇原则,弘毅给大家带来相关的无代码数据库介绍。这周就从Cell BLAST开始,一款高质量scRNA-seq数据检索/注释工具,一起来康康吧~! ... 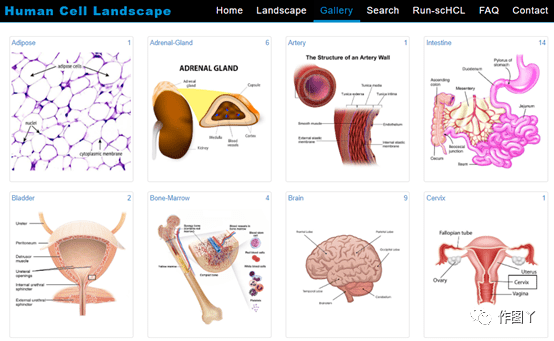 导语GUIDE ╲近几年,单细胞的研究蔚然成风,受到很多科研人员的青睐。小编为大家整理了迄今为止,最全的单细胞线上数据库,可以帮助你更准确地进行细胞注释和分析,也可以用作研究前期的数据探索。单细胞数据库分类Human Cell Landscape (HCL)Human Cell Atlas (HCA )Human Protein Atlas (HPA)SC2di...阅读全文>>...
导语GUIDE ╲近几年,单细胞的研究蔚然成风,受到很多科研人员的青睐。小编为大家整理了迄今为止,最全的单细胞线上数据库,可以帮助你更准确地进行细胞注释和分析,也可以用作研究前期的数据探索。单细胞数据库分类Human Cell Landscape (HCL)Human Cell Atlas (HCA )Human Protein Atlas (HPA)SC2di...阅读全文>>... 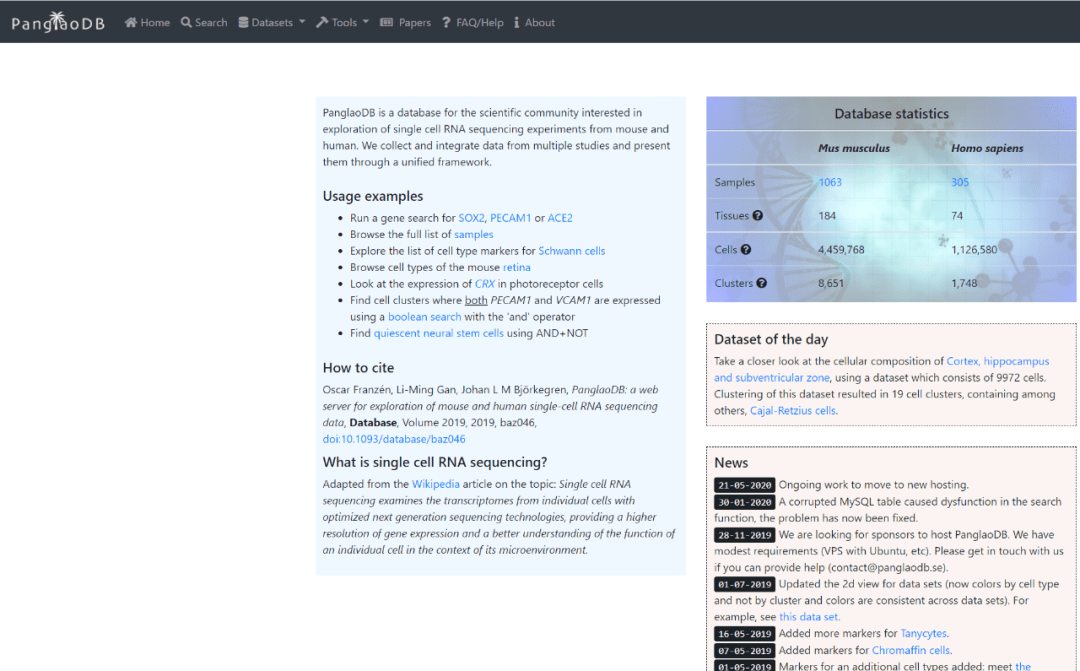 单细胞测序技术能够精确细分每个细胞独特的基因表达特征,深入研究细胞间的相互作用关系,在肿瘤免疫等方面成为人们越来越看重的技术,热度也是不断上涨。随着热度的增加,关于单细胞的文献数据也是爆炸性增长!大家在学习单细胞技术和写论文时,很多问题也是爆炸性增长!那么如何快速找到需要的单细胞数据?有哪些好用工具可以帮助我们完成日思夜想的论文?今天小编在这里挑选了一些好用且有特色的单细胞数据库分享给大家。1、P...
单细胞测序技术能够精确细分每个细胞独特的基因表达特征,深入研究细胞间的相互作用关系,在肿瘤免疫等方面成为人们越来越看重的技术,热度也是不断上涨。随着热度的增加,关于单细胞的文献数据也是爆炸性增长!大家在学习单细胞技术和写论文时,很多问题也是爆炸性增长!那么如何快速找到需要的单细胞数据?有哪些好用工具可以帮助我们完成日思夜想的论文?今天小编在这里挑选了一些好用且有特色的单细胞数据库分享给大家。1、P... 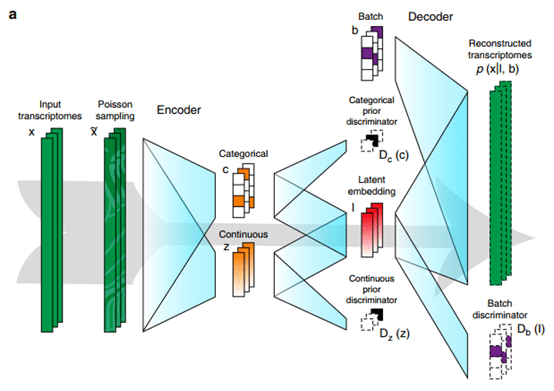 Cell BLAST作为细胞异质性研究的重要工具,单细胞转录组测序技术近年来蓬勃发展,积累了大量研究数据。Cell BLAST是一个自带高质量参考数据库的scRNA-seq数据检索/注释工具。这个网站由北京大学的研究团队研发,今年7月份相关论文发布在在《Nature Communications》:基于深度学习模型的scRNA-seq数据检索和注释的新方法Cell BLAST,以...阅读全文&g...
Cell BLAST作为细胞异质性研究的重要工具,单细胞转录组测序技术近年来蓬勃发展,积累了大量研究数据。Cell BLAST是一个自带高质量参考数据库的scRNA-seq数据检索/注释工具。这个网站由北京大学的研究团队研发,今年7月份相关论文发布在在《Nature Communications》:基于深度学习模型的scRNA-seq数据检索和注释的新方法Cell BLAST,以...阅读全文&g...  近日,美国麻省理工学院联合Broad研究所、加州大学等研究团队在 Nature genetics 上发表了题为“Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data”的研究文章。研究人员引入了 单细胞疾病相关性评分(scDRS),这是一种在单细胞...
近日,美国麻省理工学院联合Broad研究所、加州大学等研究团队在 Nature genetics 上发表了题为“Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data”的研究文章。研究人员引入了 单细胞疾病相关性评分(scDRS),这是一种在单细胞...